TFOS DEWS II - Clinical Trial Design
Gary D. Novack, PhD1,'Correspondence information about the author PhD Gary D. NovackEmail the author PhD Gary D. Novack, Penny Asbell, MD, Stefano Barabino, MD, PhD, Michael V.W. Bergamini, PhD, Joseph B. Ciolino, MD, Gary N. Foulks, MD, Michael Goldstein, MD, Michael A. Lemp, MD, Stefan Schrader, MD, PhD, Craig Woods, PhD, MCOptom, Fiona Stapleton, PhD, MCOptom
| Download this section in PDF format |
Abstract
TThe development of novel therapies for Dry Eye Disease (DED) is formidable, and relatively few treatments evaluated have been approved for marketing. In this report, the Subcommittee reviewed challenges in designing and conducting quality trials, with special reference to issues in trials in patients with DED and present the regulatory perspective on DED therapies. The Subcommittee reviewed the literature and while there are some observations about the possible reasons why so many trials have failed, there is no obvious single reason other than the lack of correlation between signs and symptoms in DED. Therefore the report advocates for conducting good quality studies, as described, going forward. A key recommendation for future studies is conduct consistent with Good Clinical Practice (GCP), including use of Good Manufacturing Practice (GMP) quality clinical trial material. The report also recommends that the design, treatments, and sample size be consistent with the investigational treatment, the objectives of the study, and the phase of development. Other recommendations for pivotal studies are a priori selection of the outcome measure, and an appropriate sample size.
Keywords: Biostatistics; Clinical trial design; Clinical trials; Dry eye disease; Regulatory affairs
(DEWS) published its report, including a report from the Clinical Trials Subcommittee [1]. In this report, which forms part of TFOS DEWS II, the previous report is updated and extended. This includes critical review of the execution of the recommendations of the previous report, the accomplishments of the previous decade, and recommendations for future clinical studies. In particular, this subcommittee believes that quality clinical studies, appropriately staged by development phase, are required for the evaluation of novel therapeutics needed by patients with Dry Eye Disease (DED).
In a companion publication from TFOS DEWS II [2], the Definition & Classification Subcommittee provided this definition: ‘Dry eye is a multifactorial disease of the ocular surface characterized by a lack of homeostasis of the tear film, and accompanied by ocular symptoms, in which tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities play etiological roles.’ For the purposes of this report, it is explicit that implied in “lack of homeostasis of the tear film” are clinical signs including corneal, conjunctival, and palpebral pathology. All drugs approved in major markets for the treatment of DED used signs as at least part of the indication (see Section 10.1).
2. Goals of the Clinical Trials Subcommittee back to top
The goals of the Clinical Trials Subcommittee are to review systematically the literature, procedures, and concepts related to clinical trials in DED. Building upon the information published in the Report of the Clinical Trials Subcommittee of the TFOS DEWS [1], an analysis of new information regarding conduct of clinical trials in DED was performed in conjunction with assessment of implementation of recommendations of the 2007 TFOS DEWS Report. This analysis included evaluation of technological advances in the field and changes in regulatory policies governing registration of new treatment options. In addition, the report makes recommendations as to best practice in clinical trials design, execution, and reporting. Finally, in the recommendations for quality clinical trial design, conduct, analysis, and reporting, reference is made to established worldwide guidance for clinical research.
3. Update on TFOS DEWS clinical trials report back to top
3.1 Progress to date in clinical trials for dry eye
The report of the Clinical Trials Subcommittee of the TFOS DEWS provided guidelines for clinical trials in general, including design, inclusion and exclusion criteria, outcome measures, sample size, randomization, data analysis, and administration of clinical trials [1]. The report recognized that both environmental trials and controlled adverse environment (CAE) trials had merit in evaluation of DED. The CAE design was judged useful for demonstration of a pharmacological effect of a treatment during monitored stress and activity. Environmental trials were judged as providing more general information about subject response to treatment on a day-to-day basis. Such recommendations continue to be relevant to the design and conduct of clinical trials.
Guidelines for clinical trials specifically in dry eye disease were based upon observations from previous clinical trials and highlighted peculiarities of clinical trials in dry eye, evaluation and outcomes parameters, and suggested desirable attributes of clinical trials in dry eye. Features to facilitate multicenter and international collaborative clinical trials were also presented in an effort to encourage international cooperation. Vagaries of DED complicating clinical trials were identified, such as the variability in concordance between signs and symptoms, fluctuations in both symptoms and signs over time as well as the possible confounding lubricant effect of control interventions.
The most desirable design for a clinical trial was recommended to be a prospective, randomized, double-masked, placebo- or vehicle-controlled, parallel-group trial. Crossover clinical trials were acknowledged to be acceptable but with the requirement that the initial treatment not cure the disease, there be no carry-over effect between periods, and all patients complete all periods of the trial. A compensatory design strategy for crossover clinical trials was suggested to randomize the sequence of the administration of test agent and control agent, so that some subjects would receive the active therapy first whereas others would receive the control therapy first. This design strategy has not seen implementation in clinical trials in dry eye, probably due to the added complexity of randomization.
The report advised that inclusion criteria should identify, based upon the mechanism of action of the proposed therapy, a potentially responsive population in which the treatment is likely to demonstrate efficacy. Inclusion and exclusion criteria should select a population that avoids or minimizes confounding variables and regression to the mean. Outcome variables should be selected consistent with the mechanism of action of the drug or intervention being tested.
The subcommittee strongly advised inclusion of biomarkers and/or surrogate markers of disease state for future trials, but recognized that validation of such surrogate markers would be needed. The outcome measures should be quantifiable with adequate accuracy and reproducibility. Measurement of the primary outcome parameter should be accomplished with a well-validated test.
Recognizing the prominent placebo and vehicle response in clinical trials in dry eye, the subcommittee recommended consideration of 1) a randomized, masked trial, in which the initiation of treatment is also masked both to investigator and subject, or 2) a withdrawal study, in which all patients initially receive active medication, followed by randomization to vehicle or active. One benefit of such a design is that all subjects receive active medication at some point in the trial, and this feature may serve to improve willingness of subjects to enroll in a well-designed trial.
Outcome analysis in a multi-factorial disease with several clinical parameters of tear film abnormality, ocular surface damage, and functional impairment was considered amenable to composite indices of disease severity. This approach has been utilized in evaluation of rheumatic disease, with consensus development of the American Congress of Rheumatology (ACR) indices (ACR20 through ACR70) that evaluate multiple descriptors of disease severity [3,4]. There has been infrequent evaluation of such composite indices in DED, and additional validated indices are needed [5,6].
The TFOS DEWS of 2007 sought to improve clinical trial effectiveness in dry eye to facilitate regulatory approval of treatments for dry eye disease, but the first drug approved by the United States (US) Food and Drug Administration (FDA) FDA in 2002 was for the indication of “decreased production of tears presumed to be due to inflammation” and the only other drug approved to treat the signs and symptoms of dry eye in the United States has been in July, 2016 [7]. (http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/208073Orig1s000ltr.pdf) Since 2007, other drugs have been approved in Japan and Europe based upon signs of dry eye disease [7].
3.2 Execution of TFOS DEWS recommendations
Few of the specific recommendations of the TFOS DEWS Report have been implemented in clinical trials designed and conducted since that report. One reason for this lack of adoption of suggested strategies is undoubtedly that such strategies require increased complexity in execution and greater expense. There is yet to be performed a randomized withdrawal clinical trial in dry eye despite the use of such strategy in other clinical areas [8-12]. The validation of reliable biomarkers of disease is an ongoing need, as is validation of proposed composite indices as outcome parameters.
The observations and refinements to the recommendations of the DEWS Report that are presented in the subsequent sections of this document will hopefully lead to future clinical trials supportive of regulatory approval and adoption of better therapies for DED. There are still significant unmet needs for management of this disease.
4. Clinical operations back to top
4.1 International conference on Harmonisation worldwide standard
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is a unique organization founded in Brussels in 1990, and now based in Switzerland. The ICH brings together the regulatory authorities and pharmaceutical industry to discuss scientific and technical aspects of drug registration. Since its inception, ICH has gradually evolved to respond to the increasingly global face of drug development. Among the more than score of guidelines is E6, the guideline for Good Clinical Practice (GCP) [13]. As stated in that guideline, GCP is an international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible. Amongst product development professionals, this 60 page guidance, as well as other ICH guidances, sets a clear standard for documented conduct of studies with respect to clinical trial design, conduct, oversight, recording and reporting while continuing to ensure human subject protection and data integrity. Standards regarding electronic records and essential documents intended to increase clinical trial quality and efficiency have also been updated. For example, if one were reading a table of the key efficacy measure in a clinical study, there is a documented audit trail that includes the statistical program that generated the table, the individual patient values in a database, and the source document for each patient's visit (ie, the first recording of the data). It also includes documentation of the investigational site that includes the investigator's credentials and training (both as an eye care specialist and for that study in particular), and the instrumentation at the site (eg, tonometer). If a photographic or video aid is to be used in a given clinical examination (which might be of value in some clinical signs for DED), it is to be documented. GCP also includes documentation of the investigational product used, including lot number, which in turn will reference information on its manufacture, meeting of release specifications, and ongoing stability and sterility. While there are other standards (e.g., for each major extramural study, the National Institute of Health creates a manual of operations), we recommend that compliance with GCP be the default standard for assurance of clinical trial validity.
4.2 Increasing standards from phase 1 – phase 3
Product development covers a range of studies – from initial studies in healthy volunteers (as appropriate), to pilot short-term, safety and efficacy studies, to pivotal safety and efficacy studies. While this report (see Section 11), as well as other reports in TFOS DEWS II [56], discuss the requirements for a pivotal study including a sample size to provide adequate power to test for clinically significant differences, such a standard is not possible for early stage studies. The reasons for this are several-fold. First, initial studies, at least with drugs and biologics, must be conscious of safety. These early studies are typically of short duration, and may escalate from low doses to higher doses. At this early stage, while the drug product still must meet Good Manufacturing Practices (GMP), it is often in limited supply with only relatively short-term stability in a pilot formulation or container-closure system. Also, typically only short-term preclinical safety studies have been completed which may not yet include all of the genotoxicity, fertility and reproduction studies. The sample size of these studies is thus small, as one would not want to expose a large number of subjects until more is known about the nonclinical and clinical safety. The magnitude of treatment effect may not be known until after initial efficacy trials are conducted. Finally, small firms sponsor many of these early stage studies in order to generate pilot efficacy data in order to obtain additional investment to conduct larger, pivotal studies.
4.3 Source documents for clinical trials
As noted in Section 4.1, part of GCP is identifying the source document for clinical data, and then assuring the integrity of the data all the way through the process. As noted in an FDA guidance, “… source data includes all information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical investigation used for reconstructing and evaluating the investigation. Access to source data is critical to the review and inspections of clinical investigations. The review of source data by both the FDA and sponsor is important to ensure adequate protection of the rights, welfare, and safety of human subjects and the quality and integrity of the clinical investigation data. Source data should be attributable, legible, contemporaneous, original, and accurate (ALCOA) and must meet the regulatory requirements for record keeping.” (Guidance for Industry: Electronic Source Data in Clinical Investigations, September 2013, http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm328691.pdf).
In previous years, source data was typically a hand-written patient chart, from which data were manually transferred to a Case Report Form (CRF) with carbonless copies, which were then distributed. The CRF was then manually keypunched into a database. With the advent of automation, today's clinical trials are often conducted with source data in electronic format. The above reference guidance provides recommendations regarding the “capture, review, and retention of electronic source data in FDA-regulated clinical investigations … ensuring the reliability, quality, integrity, and traceability of data from electronic source to electronic regulatory submission.”
4.4 Clinical supply quality
As noted in Section 4.2, the quality of investigational product (drug, biologic or device) changes throughout development. While quality products are required even at the initial stage with regard to identity, strength, quality, purity, and with most ophthalmic products, sterility, the quality level changes during development. The requirements for GMP for early stage, Phase 1 studies are provided in an FDA guidance [15]. In addition, there are standards for later stage development [16].
With respect to topical or injectable ophthalmic products, there is a requirement for sterility, as well for the containers and closures to be sterile at the time of filling and closing, and to have tamper-evident seals. Liquid ophthalmic preparations packed in multiple-dose containers should either contain an antimicrobial preservative or have a system to prevent contamination (21 CRF 200). The latter may include multi-dose, non-preserved systems as marketed widely in Europe but only recently in the U.S. and not in Japan (as of October 2016). This requirement is typically well understood and complied with by corporate sponsors of clinical trials. However, the subcommittee observed other published studies, which do not apparently provide such assurance, such as those involving multi-dose, non-preserved products in standard bottles. We caution that patient safety as well as validity of clinical trials necessitates compliance with this requirement.
4.5 Authorship standards
The International Committee of Medical Journal Editors (ICMJE), a respected group of experienced editors, created recommendations for the conduct, reporting, editing and publication of scholarly work in medical journals [17]. We encourage researchers to read this document before writing a manuscript for best practice and ethical standards in the conduct and reporting of research and other material published in medical journals. Among the topics covered are “Who is an author”, the roles and responsibilities of authors, and integrity of authors, editors and reviewers. The ICMJE also has guidelines for disclosure of conflicts of interest.
Our subcommittee, which includes section editors and editors-in-chief for several peer-reviewed journals, found that, while many clinical studies are reported at scientific meetings or in press releases, full reports are published either many years later or not at all. We interpret this finding as negative for the field. Clinical trials represent a major investment of either public or private funds, and thus we suggest an obligation to publish wherever possible. In particular, we encourage submission of manuscripts for trials with a negative outcome. Without knowledge of such trials, future researchers are condemned to repeat them. The issue of publication bias (including the underreporting of negative trials) is well known [18,19].
4.6 Clinical trial registration
In 2004, the ICMJE made a proposal for public, a priori, comprehensive registration of controlled clinical trials. Their proposal addressed the selective reporting of trials and the distortion that this created in the body of evidence in the literature. Their proposal required, as a condition of consideration for publication, registration in a public trials registry prior to the onset of patient enrollment [20]. This proposal was accepted by many of the top medical journals in the world, as well as by the Association for Research in Vision and Ophthalmology (ARVO). It was a great example of collegial responsibility amongst scientists to improve the discipline. It subsequently became law in the U.S. in 2007 (Food and Drug Administration Amendments Act of 2007, U.S. Public Law 110–85, 27 September 2007). Key to the credibility of the trial is that it be registered on a public database (e.g., www.clinicaltrials.gov) at the time of study start (defined as within 21 days after the first patient is enrolled). Registration also is law in other countries and other public registries exist (e.g., www.umin.ac.jp/ctr/index-j.htm; www.clinicaltrials.jp/user/cte_main.jsp; www.who.int/ictrp/en; www.allerganclinicaltrials.com) The most commonly used registry is www.clinicaltrials.gov, which is hosted by the National Institutes of Health (NIH) and FDA, although other countries and organizations, as well as some pharmaceutical firms have set up their own websites (e.g., World Health Organization, http://www.who.int/ictrp/en/; Japan, http://www.umin.ac.jp/ctr/, http://www.who.int/ictrp/network/jprn/en/, http://rctportal.niph.go.jp, https://dbcentre3.jmacct.med.or.jp/jmactr/Default_Eng.aspx, http://rctportal.niph.go.jp/link.html, http://www.clinicaltrials.jp/user/cte_main_e.jsp, Australasia, http://www.anzctr.org.au, Glaxo SmithKline, http://www.gsk-clinicalstudyregister.com/). The passage of the 2007 law resulted in a dramatic increase in the number of clinical trials registered. However, it seems that not all researchers are following regulations regarding reporting of results, which in turn is leading to increased reporting requirements by NIH [21-23].
During the conduct of a study, access to data is very limited, consistent with GCP. Similarly, during the predefined statistical analysis, the data are closely held. In some cases, the results of a study may be considered a material event for the sponsor, and thus release may affect the stock price. A summary of the data may be provided in a press release or at a meeting. However, once the evaluations are complete and the planned analyses conducted, some feel that, in a spirit of transparency, raw data, with patients de-identified, should be available to others, albeit with some controls in place. With respect to studies supported by government funds, data from large, extramural studies conducted by the National Institutes of Health (NIH) may be available upon request, e.g., Collaborative Longitudinal Evaluation of Ethnicity and Refractive Error (CLEERE) Study. These requests are reviewed by a team of the study members for merit. The NIH also has a policy on sharing human and model-organism genotypic and phenotypic data, including embargo periods to ensure that data producers have sufficient time to analyze their results [24]. In the field of astronomy, data obtained from observatories (both ground- and satellite-based) are released at a given period after they are recorded. For research sponsored by the private sector, there are concerns about confidentiality. One example of data sharing has been provided by Glaxo Smith Kline (GSK). Since May 2013, investigators have been able to request access to de-identified patient-level data from clinical trials sponsored by GSK, subject to review and oversight by an independent review panel (https://clinicalstudydatarequest.com/Default.aspx). Of the 77 complete requests received in the first year of this program, nearly 100% were approved. Results of these analyses are not yet available [25]. The RIAT movement (Restoring Invisible and Abandoned Trials) [26], resulted in the independent re-analysis of a trial of an anti-depressant in adolescents from 2001 with greatly different results [27,28].
In January 2015, the US Department of Health and Human Services (HHS) proposed a rule to implement the requirements of the Food and Drug Administration Amendments Act of 2007 (FDAAA), to require public sharing of summary data from certain clinical trials of FDA-regulated drugs and devices [29]. Just recently, the International Committee of Medical Journal Editors (ICMJE) proposed requirements to help meet the data-sharing obligation [30]. Some journals, including the New England Journal of Medicine [31], and Science [32] have adopted aspects of these proposals. Importantly, data sharing not only prevents a repeat of mistakes in subsequent studies, but also adds to the understanding the natural history of treated and untreated or vehicle-treated disease and a measure of temporal variability in signs and symptoms which informs effect size and aids sample size estimation in subsequent clinical trials. This issue was reviewed by Wald (2017) [33].
4.7 Standards for well-controlled studies
In 1962, the U.S. Congress passed the Kefauver-Harris amendments to the Federal Food, Drug & Cosmetic Act. That law requires approval of a new drug be based upon evidence of effectiveness which is based on adequate and well-controlled clinical studies conducted by qualified experts. The definition of an adequate and well-controlled study was subsequently defined in 21 CFR 314.126. These attributes are used by the U.S. FDA and, presumably, other regulatory bodies, in their consideration of the quality of a study submitted in support of an application. The subcommittee recommends that researchers consider these attributes at the outset of clinical trial design and in selecting sites for the conduct of a clinical trial. The items described are summarized as:
(1) Clear statement of the objectives of the investigation.
(2) Uses a design that permits a valid comparison with a control to provide a quantitative assessment of drug effect.
(3) Placebo, (ii) Dose- comparison, (iii) No treatment (iv) Active (v) Historical control.
(4) Method of selection of subjects provides adequate assurance that they have the disease or condition being studied.
(5) Method of assigning patients to treatment and control groups minimizes bias.
(6) Adequate measures are taken to minimize bias on the part of the subjects, observers, and analysts of the data.
(7) Methods of assessment of subjects' responses are well defined and reliable.
(8) There is an analysis of the results of the study adequate to assess the effects of the drug.
(9) The test drug is standardized as to identity, strength, quality, purity, and dosage form to give significance to the results of the investigation.
In 1996, a group of clinical trialists, editors, and others, published an article proposing a checklist for improving the quality of reporting of randomized controlled trials [34]. In its current form, this CONsolidated Standards Of Reporting Trials (CONSORT, http://www.consort-statement.org/) statement consists of a checklist and flow diagram for reporting a randomized controlled trial (RCT). It is required by many of the top medical journals, including JAMA and The Lancet. The CONSORT statement has a number of similarities to the items described in 21CFR314.126. A criticism of the CONSORT statement is that it is usually applied by researchers after the study is completed, and a manuscript is being prepared, rather than as the study is being designed [35].
4.8 Ethics and data safety committees
Studies involving human subjects require approval and oversight by an Institutional Review Board (IRB) or Independent Ethics Committee (IEC) in order to safeguard the rights, safety, and well-being of all trial subjects. Special attention is to be paid to trials that may include vulnerable subjects. Additional details on the role of IRB/IECs are provided in Section 3 of the ICH E6 guidance. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Addendum_Step2.pdf.
Some studies also employ a Data Monitoring Committee (DMC). Ellenberg states that the purpose of a DMC is to advise the study sponsor regarding the continuing safety of current participants and those yet to be recruited, as well as the continuing validity and scientific merit of the trial to protect the safety of trial participants. It has some, but not 100% overlap with the responsibilities of the IRB/IEC [36] There is relatively little in FDA laws or regulations regarding the DMC, and they are mentioned only briefly in the ICH guidelines. The most current US regulatory guidance is from 2006 (http://www.fda.gov/downloads/regulatoryInformation/Guidances/ucm127073.pdf). Typically, small studies at early stages of product development do not require a DMC nor support the logistics required. Most studies of pharmacological dry eye treatment do not require a DMC, as patient safety issues are not of a magnitude to require unmasking by a third party. Further, the treatments, at the time of study conduct, are typically either an investigational product or its vehicle, so that even if one treatment were substantially more effective than another, it would not affect the treatment of other patients with the disease [37].
5. Special issues for dry eye trials back to top
5.1 Low success rate for pharmacotherapy of dry eye disease
In addition to providing information on general issues on designing and conducting quality clinical trials, the report includes discussion of special issues in trials of treatment for DED. This is of particular interest to patients, researchers, and clinicians given the large number of novel agents evaluated in clinical trials, yet the relatively low number of pharmacotherapies approved [38]. The subcommittee received extensive input on the reason(s) why so many clinical studies failed. After careful review of the literature, while there were some suggestions, there is no one obvious reason for these failures, other than the lack of correlation between signs and symptoms in the disease. Compliance with good trial design, as recommended in this report, should lead to improvements in outcomes.
5.1.1 Commonly encountered problems
As noted in the introduction to TFOS DEWS II, the approval of new therapeutic agents for the treatment of DED has been very slow and infrequent. At the time of writing, only two therapeutic drugs, cyclosporine A (indicated for the improved production of tears in those with reduced tear production thought to be due to inflammation) and lifitegrast (indicated for the relief of signs and symptoms of dry eye) have been approved for treatment of DED in the U.S., despite many clinical trials. The exact number of failed trials in this area is not clear, as many have died quietly without fanfare. A similar drug, cyclosporine A with a different formulation, was approved in the European Union (EU) in 2015. There are several approved drugs in Japan, but these have not been approved by the regulatory agencies in most other major countries. It is possible that the tested yet failed formulations were insufficiently active in controlling the disease. But, what is more likely, is that the design of clinical trials and an incomplete understanding of the disease pathogenesis and development along with regulatory requirements based on earlier understanding of the disease have contributed to the failure rate. The Report of the Inaugural Meeting of the TFOS i2 has a list of failed trials, which utilized corneal staining as a primary endpoint (Table 10) [39]. Recent advances in understanding indicate that corneal staining is a late finding in the development of disease and that it, like most other objective tests, is variable over time owing to the instability of the tear film, a hallmark of DED [6,40]. Other confounding pitfalls seen in clinical trials for therapeutic agents in DED are discussed here. A better understanding of these areas, particularly inclusion of molecular markers, should lead to better outcomes and success in the development of clinically useful therapies.
5.1.2 Definitional problems
As noted in the recently published TFOS i2 Report, there is widespread disagreement throughout the literature, both on the definition and diagnosis of dry eye disease (DED) [39]. The disease entity is viewed as a disease, a constellation of symptoms, a list of different ocular surface conditions, or a specific condition associated with other conditions (e.g., aqueous tear deficiency and meibomian gland dysfunction (MGD). With this broad conceptual approach, it is impossible to compare results of clinical trials. There is a large body of literature supporting the concept of a functional lacrimal unit consisting of the main and accessory lacrimal glands, the tear fluid, the cornea, conjunctiva and meibomian glands, and, more recently the tear ducts, all of which are connected by a neural network. This nerve-based unit is responsible for maintaining homeostasis of the ocular surface and tear film, which allows clear vision and for maintenance of the constituents of the ocular surface, between blinks, in response to environmental stress [41].
The original DEWS report included a mechanistic division of DED based on which major contributors to the tear film are primarily affected; the lacrimal glands or meibomian glands of the lids. These two major subtypes of disease are termed aqueous deficient dry eye (ADDE) and evaporative dry eye (EDE), respectively. The most common of the two is EDE [42]. These two subtypes can present individually but are related and elicit compensatory responses, which affect tear stability and ultimately lead to a combined form of DED [40,43].
5.1.3 Effects of bilaterality of DED in design of clinical trials
Dry eye disease usually presents as a bilateral disease although there can be differences in disease severity between eyes. Despite the preponderance of a bilateral presentation of DED, many clinicians and investigators treat each eye as a separate unit with significant effects on clinical trial design and results. It is well established that normal eyes do not function as independent units but rather communicate with each other with effects on the function of the other, examples might include a sympathetic response to accommodation and contact lens-induced corneal swelling [44]. This inter-eye signaling has been demonstrated in reports describing unilateral quiescent herpetic keratitis in which the contralateral eye without herpetic infection has been found to exhibit a decrease in corneal sensation and aqueous tear production [45,46]. The presumed normal eye has also been shown to have significantly higher discomfort levels, visual symptoms, reduced tear breakup time, and an increase in tear osmolarity with increased inter-eye differences compared with normal controls [46,47]. A recent study has demonstrated that, in patients with unilateral infectious keratitis, there is a sympathetic immune response with effects on sub-basal corneal nerves and sensation and an increase in dendritic cell density in the contralateral eye [48].
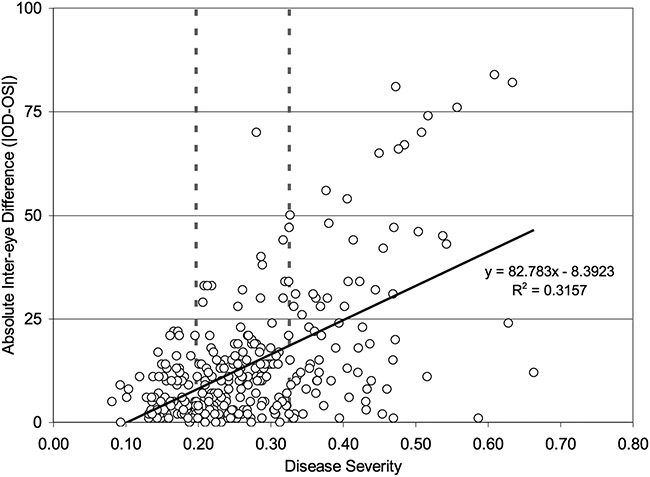
A hallmark of DED is an unstable tear film, which is associated with variability in objective measures of DED [40,49], In a study of repeated measurements of tear osmolarity and other signs over a 3-month period, the more severe eye shifts from one eye to the other from 19% to 4% of the time [50]. Also, an inter-eye difference greater than 8mOsm/L, which increases with progression of the disease is an additional hallmark of the tear film instability and severity of disease (Fig. 1) [40,51]. This inter-eye difference decreases with effective treatment of DED [5,52,53]. The phenomenon where the variability of one variable (in this case, the interocular difference in tear osmolarity) is unequal and depends upon another variable (in this case, the disease severity) is called heteroscedasticity.
With the unfortunate experiences in many failed dry eye clinical therapeutic trials, it is possible that this loss of valuable information may have played a role in the failure to demonstrate efficacy. The two eyes do not seem to function as independent variables in DED but rather as two parts of a single unit (the patient), which should be treated as such [54]. Typically, one selects a study eye (usually the worse eye, or an average of eyes) per study subject, as eyes are typically NOT independent, and one observation per patient is used [55]. However, in this case, measuring both eyes, and taking the interocular difference in tear osmolarity as a measure for efficacy may be of value. Some of the authors have experience with planning of treatment studies under a U.S. Investigational New Drug (IND) exemption using this interocular difference. Note that this approach still results in one observation per patient, which is the appropriate statistical sample.
5.1.4 Selection bias
The selection of diagnostic criteria to determine which patients are selected to enter a clinical trial plays a pivotal role in determining outcomes. In the absence of a widely accepted “gold standard” for identifying subjects with DED, the selection of a group of characteristics for inclusion will be critical. As mentioned above, definitional decisions as to what constitutes DED should be clear (e.g., are these inclusions definitive for ADDE only, or EDE only, or do they relate to a definition of DED which includes both). Selection of endpoints, which include those markers that are in the inclusion criteria, favors their therapeutic outcomes versus those endpoints not in the definition and for which many subjects would enter the study with normal values (which cannot be improved upon). This bias has occurred in recent papers leading to unsupported conclusions [54].
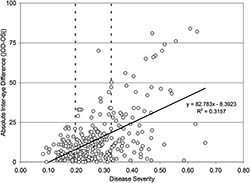
Fig. 1 Relationship difference in inter-eye osmolarity versus Dry Eye Disease severity (Reproduced with permission from Lemp et al. Am J Ophthalmol 2011; 151:792–798 [40]).
Another pitfall to be avoided is the inclusion of endpoints collected on limited pre-study visits owing to the tendency for regression to the mean. This occurs when subjects are recruited based on endpoints which are abnormally high for inclusion but which vary over time with or without treatment. There is a large likelihood that a sample at a subsequent time point may result in a lower value, attributable not to therapeutic effect but rather to the natural cycle of variability over time. This effect can be minimized by having entrants to a study qualify with readings at more than one visit before inclusion. This is discussed further in the TFOS DEWS II Diagnostic Methodology report [56].
5.1.5 Spectrum bias
This refers to differences in the features of different populations that would influence sensitivity and specificity (see below). In therapeutic trials, most often this occurs when inclusion criteria limit the study cohort to those with a particular level of disease severity, and the results are generalized for more or all groups. This is a fairly common bias but sometimes is justifiable in therapeutic trials that are seeking subjects with sufficiently abnormal endpoints to provide a dynamic range sufficient to demonstrate improvement (e.g., corneal staining). As long as this bias is recognized and the results are not generalized as applicable for all levels of severity, this is perfectly acceptable. This is discussed further in the TFOS DEWS II Diagnostic Methodology report [56].
5.1.6 Specificity and sensitivity usage in identifying study subjects
It is widely recognized that the terms sensitivity and specificity have importance and are frequently referred to in reports of clinical trials [52]. In a field in which there is not a consensus on the important and practical diagnostic characteristics that identify subjects with DED, there is a tendency to employ diagnostic criteria for judging a subject as having DED or not, which may favor changes in a specific marker (e.g., corneal staining) which is also an outcome measure.
Sensitivity refers to the proportion of patients with the disease who have a positive test result. Specificity refers to the proportion of normal subjects with negative test results. As discussed in the previous DEWS report, it is usually considered more important in assessing results of a therapy in a non-fatal disease such as DED, not to miss subjects with disease; this elevates the importance of results on sensitivity and positive predictive value (PPV), the probability of those with a positive test result actually have DED. Attention to the defining aspects of classification of subjects is a critical factor in the interpretation of clinical trial results. This is discussed further in the TFOS DEWS II Diagnostic Methodology report [56].
5.2 Study design
The clinical study design features of double-masking and randomization of treatment assignment, introduced in the 1930s by Harry Gold [57] as cited by Reidenberg [58], are considered a standard for high quality research. There are a host of issues to consider when designing a clinical study. These include the investigational agent and control(s), study subject inclusion and exclusion criteria, design (e.g., parallel vs. crossover vs. paired-comparison), selection of efficacy and safety outcomes, and statistical issues. Some of these issues were covered in the earlier section on previously failed clinical trials (Section 5.1). In this section, key design elements as they apply to study of treatments for DED are described. Details on study design, including randomization and masking may be found in the ICH E6 and E9 guidance. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm073122.pdf, http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf.
5.2.1 Inclusion/exclusion
An ideal study would consist of inclusion and exclusion criteria that provide subjects with a homogeneous disease status that is responsive to the mechanism of action of the intervention (study drug) and still allows generalization of the study findings to the larger population of patients who suffer from dry eye. Inclusion and exclusion criteria also are used for ethical considerations and to protect the validity of the study [59]. Due to ethical concerns, patients from vulnerable populations typically are excluded, and subjects must have the ability to provide informed consent. Most dry eye studies are limited to patients who are 18 or older [60–62].
5.2.1.1 Inclusion criteria
Inclusion criteria are used to ensure that the subjects can comply with the study protocol and have dry eye with a homogeneous disease status. Studies typically use the signs and symptoms outcome end points to determine whether subjects qualify for the study [63,64], and outliers on either end of the measurement spectrum are not included since they have the potential to cause regression to the mean or may not be responsive to the study medication [60,62,65,66].
Measures of ocular surface staining and symptom surveys, such as Ocular Surface Disease Index (OSDI), equally weighted, are frequently used to determine if subjects have qualifying disease severity [67]. Other variables include measures of tear production (Schirmer tests), normal lid positioning, and best-corrected visual acuity [64]. By enrolling subjects who report the recent use of artificial tears (within 30 days), the study includes subjects who are symptomatic enough that they seek treatment for dry eye and also subjects who can self-administer eye drops, which is the method of drug delivery for most dry eye studies [62].
5.2.1.2 Exclusion criteria
Individuals who are pregnant, nursing, or could become pregnant, are typically excluded from dry eye studies [60,62]. Patients with known hypersensitivity to the study medication, and, often patients who have previously used the study medication topically or systemically are also excluded [60]. Many studies will exclude people who have ocular and/or systemic conditions that could confound the results (See Section 5.3), and discretion is often given to the investigator and/or medical monitor to determine whether the condition has the potential to distort the relationship between the study drug and the outcomes [60,62]. Potential study subjects often are excluded if they take medications known to influence the outcome parameters [60,62]. However, some studies will allow enrollment of subjects using concomitant medications as long as they are on a stable dosing regimen that will continue throughout the duration of the study [60]. Patients also are excluded if they have had recent ocular surgery or are planning to have eye surgery during the study period [62]. Other possible exclusion criteria are: prior ocular surgical procedure, clinically significant ocular trauma, use of contact lenses, best corrected visual acuity worse than a given limit, hypersensitivity to any of the ingredients, current or recent use of punctal plugs, or use of another investigational agent within the past 30 days.
5.2.2 Randomization and masking
The randomized clinical trial (RCT), if possible, continues to be the highest level of demonstration of the therapeutic value of a product [68]. As with randomization and use of comparators, the reason for masking participants from what treatment they have received is to control bias. For this to have an optimal effect, masking should be provided for both investigator and patient volunteer, in other words ‘double masking’ [69]. Where double masking is not possible, the reasons should be acknowledged a priori in the protocol, and in other study reports [70]. Details on randomization and masking procedures may be found, once again, in the ICH E6 and E9 guidance.
5.2.3 Selection of controls
Clinical comparison of the “active” with a vehicle, placebo, or other “inactive” is a challenge for the design of a trial in DED. As noted previously (Section 5.1), there are relatively few approved pharmacotherapies for dry eye for selection as an active control.
Placebos are extensively used as part of investigating the performance of investigational agents. Hrobjartsson and Gotzsche (2010) state that the placebo should be selected with great caution, especially in clinical trials that investigate conditions resulting in pain [71]. Given the importance of symptoms in dry eye disease, trials of new agents would qualify as areas of concern [71]. With respect to negative controls, a true “vehicle” is the same as the Active formulation, minus the Active Pharmaceutical Ingredient, perhaps slightly adjusted for the impact of the active on pH, tonicity, viscosity, etc. Because, in most cases, the vehicle is effectively an artificial tear, the dynamic range for clinical differentiation is compressed.
Foulks expanded on this concept in a review in the early 2000's [72]. In contrast to a placebo, a “nocebo” is a maneuver, instruction, or substance that inherently does not worsen the condition nor provoke an adverse event, but which the patient interprets as aggravating the condition being treated or producing an unwanted adverse event. Foulks hypothesized that one possibility for the large placebo response seen in DED trials is that such a placebo response may be due to improved compliance in using the prescribed medication by patients enrolled in a clinical trial (i.e., a “Hawthorne Effect”) [73,74]. Another consideration is that, if patients prior to enrollment in a clinical trial were using preservative-containing lubricants or other topical medications, and then discontinued their use for the trial, improvement in symptoms and ocular surface staining may be due to recovery from adverse effects of the preservative. If there is not an adequate washout period prior to institution of randomized therapy, improvement can be seen in all treatment groups [59,75]. This feature may be planned in a withdrawal trial of a therapy (See Section 7.7.4).
Given the variance of the response, the sample size may become impractically large and/or the magnitude of difference between active and vehicle clinically irrelevant (See Section 11).
5.2.4 Qualifying phase
Frequently a clinical trial design will include a washout and/or qualifying period. With the approval of DED therapeutic products worldwide [7], many DED patients may be using a pharmacological therapy or punctal plugs prior to entrance into a clinical study. The authors suggest that a washout period from these active agents be employed. However, there is inadequate information to propose an evidence-based recommendation for the duration of this washout.
A number of proposals for run-in periods to address specific issues in trials of DED, including a variable period to enhance masking were proposed in the early 2000's [59]. Examples in two studies are (1) a run-in period on a standardized, unmedicated, unpreserved over-the-counter lubricant [76] or (2) actual use of the product under investigation for participants to demonstrate a positive response [76–78].
5.3 Confounding factors
Confounding is often referred to as a “mixing of effects” wherein the effects of the exposure under study on a given outcome are mixed in with the effects of an additional factor (or set of factors) resulting in a distortion of the true relationship. In a clinical trial, this can happen when the distribution of a known prognostic factor differs between groups being compared [79]. If the confounding variables are equally distributed between study groups, then the distortion resulting from confounders is minimized. Practically, this can be difficult to accomplish given that dry eye is a heterogeneous disease process, with many extraneous variables that individually may have a range of effects on the signs and symptoms of dry eye; moreover, these distorting effects can be difficult to grade or measure [59,79]. Even if the effects of the confounding variables can be accurately measured and the confounders are distributed equally between study groups, the (inverse or direct) association of the confounders on the study groups could still make it more difficult to measure a statistically significant difference between study groups. For instance, more subjects may be required in a study to account for the inverse or direct association of the confounder. Therefore, it is important for clinical trials, particularly for the treatment of dry eye, to recognize and control for extraneous confounding variables [80].
Examples of potential confounders include demographics, anatomical pathological or post-surgical changes, ocular or systemic diseases, topical or systemic medications, and use of ocular devices [59]. Demographics such as age and sex are known to affect dry eye [42,81,82]. Other confounding factors may be geographical in nature. For example, latitude affects study sites in a multi-center study differently with respect to diurnal hours, as well as seasonal pollen exposure. Different locations may also have different humidity or air pollution, which may affect DED.
Lid abnormalities such as ectropion, paralysis, and Graves eye disease can result in exposure keratopathy [83]. Misdirected lashes and entropion can result in corneal staining, which is a common outcome measure of most dry eye studies. Meibomian gland dysfunction and rosacea can result in signs and symptoms of dry eye that are not responsive to dry eye interventions that aim to increase aqueous tear production [84]. It is particularly difficult to control for MGD given that it presents as a spectrum of severities and that many patients with aqueous tear deficiency also have condition [85]. Previous eyelid surgery such as the removal of lid tumors can influence the contour of the lid margins, interfering with the normal lid/tear resurfacing mechanism and may possibly cause lid wiper epitheliopathy [83].
Corneal and ocular surface diseases often influence the signs and symptoms of dry eye. Ectasias typically present with prolate corneas with central corneal staining, which is independent of aqueous tear production [81,85,86]. Multiple conditions can result in a neurotrophic cornea or corneal neuropathies. Corneal refractive surgery (e.g. LASIK) is associated with neurotrophic corneas and with dry eye [87]. A history of viral keratitis, such as herpes simplex virus (HSV), can also result in neurotrophic corneas and corneal staining [88]. Ocular surface tumors, elevated pterygium or pingueculum cause irritation and dellen formation. Actively chronic or acute infections, such as viral keratitis, HSV/herpes zoster virus (HZV), or conjunctivitis can present with ocular symptoms and corneal staining [89].
Active disease, or a history of systemic diseases, can influence dry eye study outcomes and can be problematic if their effect is not stable. Many autoimmune conditions result in dry eye, and these include rheumatoid arthritis and graft-vs-host disease [59].
Concomitant systemic and ophthalmic medications can influence dry eye study parameters. Systemic medications that have reportedly affected dry eye include steroids, immunosuppressants, parasympathomimetic agents, antihistamines, and others. Local therapies include glaucoma medications, antimicrobial drops, cyclosporine, autologous serum tears, and others [49,90,91].
Use of ocular devices such as contact lenses and punctal plugs are respectively inversely and directly associated with dry eye outcome parameters [92,93]. Contact lens wearers report increased dryness symptoms compared with non-wearers [94]. Punctal plugs are a treatment for dry eye and can improve the symptoms and signs of dry eyes, but there is a risk that the plugs may not be retained throughout the study period, which could introduce additional confounding effects [93].
Compliance with treatment regimens can be difficult to predict, detect, and control for in a dry eye study [59]. Confounding bias would exist if one treatment group is less compliant with a medication [79].
5.4 Treatment adherence and performance
C. Everett Koop, M.D., former U.S. Surgeon General, said, “Drugs don't work if you don't take them”. Forty years ago, Blackwell published a review article on treatment adherence (also called compliance) in which he proposed that adherence was positively related to symptoms and negatively related to the drug-induced adverse events [95]. Subsequently, studies using electronic monitors found relatively poor adherence, including poor persistence, with many chronic diseases – be they either symptomatic (e.g., epilepsy) or asymptomatic (e.g., hypertension) [96]. A myriad of types of adherence failures exists, including missed doses, doses taken at the incorrect time, and lack of persistence. Thus, by analogy, patients' adherence with treatments for DED may also be less than optimal. There is the additional concern that a large proportion of patients have problems instilling eyedrops [97]. In the interpretation of clinical trials, researchers must understand that adherence of study subjects may be variable. Under-dosing may result in lower efficacy, and over-dosing may result in more safety issues. These may be ascribed inappropriately to the drug. Also, adherence in clinical trials may not be representative of use in clinical practice (e.g., the previously mentioned “Hawthorne Effect”). It may be that an objective measure of treatment adherence should be incorporated into clinical trials. While studies with systemic medication may assess drug levels in the blood, in ophthalmology it is rarely possible to measure drug levels in the target organ [98]. Prescription refill rates, or pill counts (or the ophthalmic equivalent, weighing bottles) have also been used, but they do not record the time at which the medication was taken, and are highly variable. Thus, electronic devices which have been developed such as the Medication Event Monitoring System (MEMS) device (Aardex, WestRock, Richmond VA), can be used with ophthalmic products using the “bottle within a bottle” method [99].
To date, primarily pharmaceuticals have been explored for treatment of DED. These new drugs (either new chemicals and/or new to be used for ocular treatment) use traditional clinical trial design: double-masked randomized controlled trial (RCT) to ask one primary question [59,64]. Medical device and surgical interventions are typically not approached with traditional RCT, given that masking is often not possible for either the subject or the evaluating clinician, and because determining what would be an appropriate control is less straightforward than it is in pharmaceutical trials. For device trials, controls can be sham surgery, or more commonly “masking” of outcome assessors, duplicate assessment of outcomes, and use of objective outcome measures [100]. Selection of an appropriate negative control for a medical device trial may be challenging [101]. In addition to trial design, the regulatory approach for devices is quite different.
Some of the following issues get to the core of clinical trials: why is the trial being done? Typically, a sponsor is looking for regulatory approval to market the treatment for a specific population and for a specific purpose. Given that most companies are indeed focused on regulatory approval, one needs to understand that the roadmap for approval can be very different for countries around the world. In most countries, the approval process for medical devices is very different than that used for drugs and biologics. This is in contradistinction to the patient, who is relatively agnostic as to the nature of their treatment, and the clinician, who wants the largest inventory of treatments available for each patient. Depending upon the approval process, some medical devices are approved based upon their similarity to a previous device (“predicate”), and compared to essentially historical, rather than concurrent controls, or to a preset standard. Other products may require concurrent controls. By their nature, it is challenging to mask patient and clinician to medical devices. For a limited number of products, a “double-dummy design” is possible, wherein each subject receives both the investigational device and the control – each treatment group receiving active of one treatment and a “placebo” of the other treatment.
A number of controlled studies have been conducted for medical devices and surgical methods to treat DED with a wide range of study designs [102–117]. The place for these treatments in the armamentarium of DED therapy is covered in the TFOS DEWS II Management and Therapy report [14].
The development of devices is very different from that of drugs. In addition to trial design, the regulatory approach for devices is also quite different from that for drugs and biologics in most countries [118]. The process for approval is often very different for the European Union, USA and Asia, depending upon how the treatment is categorized by the regulatory agency [7,39]. Overall, regulatory agencies try to weigh the risk versus benefit of the new intervention. Review of peer reviewed publications on devices for DED treatment highlights the difficulty of evaluating efficacy and safety in device trials [102–112,114–116].
7. Efficacy: clinically relevant differences back to top
In statistics, a significant difference is simply a difference that is unlikely to be caused by chance and has a mathematical basis for such a claim. In every day health care, a difference may be statistically significant based on a numerical value. Yet, it may, at the same time, be of little or no importance to the health or quality of life of patients afflicted by a certain disease [119]. The concept of a ‘‘clinically important difference’’ has been first described by Jaeschke et al. as “the smallest difference in score in the domain of interest which patients perceive as beneficial and which would mandate, in the absence of troublesome side effects and excessive cost, a change in the patient's management” [120]. The minimum clinically important difference (MCID) is a threshold value for such a change. Any amount of change greater than the MCID threshold is considered to be meaningful or important. A simpler definition was later proposed by Stratford et al. as ‘‘the smallest change that is important to patients’’ [121]. In general, the estimation of a clinically relevant change of a sign or a symptom for a treatment that is tested in a clinical trial, considering the subtype of dry eye, helps interpret intervention effects and allows the investigator to use them with more confidence as an end point in clinical trials.
7.1 Outcome measures
The correct diagnosis and classification of a disease as well as the choice of the parameters to monitor disease progression or therapeutic response is essential for the success of a clinical trial. Dry eye is defined by several signs and symptoms, namely discomfort, visual disturbance, tear film instability, potential damage to the ocular surface, increased osmolarity of the tear film, and inflammation of the ocular surface [2,40]. When the investigator makes a diagnosis based on multiple signs, there must be consideration of the sensitivity of therapeutic response, temporal variability, possible overlay/interference of the presented symptoms with other ocular irritations, environmental influences, and the ideal distribution of each sign. This is discussed further in the TFOS DEWS II Diagnostic Methodology subcommittee report [56].
This situation is further complicated by the fact that signs and symptoms are often poorly associated in dry eye disease, which makes assessment of severity as well as choosing the right outcome measures particularly difficult. This has to be considered in clinical trial design [51,122]. Another parameter to consider when designing a clinical study is the order in which the different assessments should be performed. In a sequence of assessments, tests should be performed in an order from the least to most invasive. The TFOS DEWS II Diagnostic Methodology subcommittee report includes further detail on the recommended order of testing [56].
Tear osmolarity should be evaluated first (with no drops having been administered for at least 2 h before sampling). Slit-lamp examination assessing tear break-up time with fluorescein (least recommended), and punctate epithelial erosions of the cornea with fluorescein followed by conjunctival staining patterns evaluation with lissamine green seems to be an optimal order. However, in protocols using non-invasive tear break up testing, which would be the preferred approach, this should be measured prior to tear osmolarity [56]. Schirmer testing may be performed subsequent to staining, since contact of the Schirmer strip against the conjunctiva can cause ocular surface staining, the Schirmer test without anesthesia may be deferred until this time, but if the Schirmer strip is to be used for collection of analytes from the tear at an earlier time, the induced ocular surface staining must be ignored in grading ocular surface staining. Other invasive tests (e.g., impression cytology, tear collection, confocal microscopy) should be planned at different times to avoid changes of the ocular surface and/or problems related to the use of vital stains. Tear sampling for analytes using Schirmer strips should not be performed before measurement of tear breakup time. If it is necessary to perform both tests on the same day, NIBUT, ideally with an infra-red light source, should be performed first.
7.2 Classification of disease severity
The original Delphi panel reached a consensus that “severity of disease should be the primary determinant for the therapeutic strategy chosen [123]. The following DEWS dry eye severity grading scheme gave broad categories and a few numerical breakpoints on the path to severity, but did not address conflicts among signs [124]. Still to date, there exists no consensus on a gold standard measurement to assess disease severity in dry eye [125]. When evaluating correlation of different signs in dry eye, correlations seem possible in small subsets of patients [126], while in large cohorts, with different underlying etiologies, signs tend to respond independently from each other, as shown, such as described in a study by Huang et al. where tear protein markers were correlated with different severity groups of dry eye based on corneal staining and OSDI score. In these groups, Schirmer score and tear osmolarity showed poor correlation with disease severity [127]. It has to be noted that corneal staining is a relatively late manifestation of DED and has little pertinence in mild to moderate disease, which demonstrates the importance of choosing the appropriate markers for classifying certain groups and stages of dry eye. Also, tear osmolarity measurements in that study were not conducted in accordance with the FDA cleared guidance for testing, limiting the clinical accuracy. Nevertheless, the study shows the limited value of single signs to classify disease severity especially in early stages, when disease expression can be very variable and compensatory mechanisms may transiently alleviate the effects of environmental stress [6].
Composite indices involve mapping a series of signs onto a common basis through normalization or ordinal ranking, followed by a weighted summation of the constituent signs. One of the main benefits of any composite approach is that random temporal variability is dampened across multiple signs [63,128,129]. An intermediate step between a multiple composite and a single efficacy value is a co-primary efficacy endpoint. This was used in the evaluation of Ikervis® (cyclosporine emulsion 0.1%), where the co-primary endpoint was a categorical (yes/no) measure. In order to be considered a success, patients needed to have an improvement in Corneal Fluorescein Score of 2 units or more (Oxford scale of 0–5) and an improvement by 30% or more from baseline in Ocular Surface Disease Index (OSDI) at 6 months. The product did not meet this co-primary endpoint, but was approved based upon Corneal Fluorescein Score improvement alone (continuous analysis, −1.05 vs. 0.82 units, p = 0.009) [130]. Simply stated, the dampening achieved in a composite or a co-primary analysis also dilutes a possible treatment effect. Like any novel method used to evaluate a novel therapy, any method should be validated against a positive control. As already noted, there are few approved pharmacotherapies to use in such validation. For example, Ikervis® (cyclosporine emulsion 0.1%) was approved only for “severe keratitis,” which in that scenario meant corneal staining.
In a study by Sullivan et al., a composite score of the 7 features of DED (osmolarity testing, Schirmer test without anesthesia, TBUT, corneal staining, meibomian gland scoring, conjunctival staining, OSDI) was formed and used as the standard against which the individual tests were judged. Tear osmolarity displayed a linear relationship to the composite scores and was found to be the single best marker of disease severity across the normal, mild/moderate, and severe categories [6].
It has also been recommended that methods and treatments be evaluated using quantitative biomarkers, rather than clinical signs and symptoms (See Section 7.6) [125].
A new method to assess the severity of DED based of the number of standard deviations from an appropriately selected healthy population is further described in the TFOS DEWS II Diagnostic Methodology and Tear Film subcommittee reports [56,131].
7.3 Monitoring therapeutic response
Successfully determining response to a specific treatment in a clinical trial is key, and it has been shown that outcome measures should be selected carefully depending on the treatment since sensitivity of markers or signs differ considerably depending on the therapy being tested [51]. Outcome measures are critical in developing the trial design especially for determining efficacy. The trial design is best when the outcome measures are in line with the expected mechanisms of action of the treatment being studied. Also key is the ability to adequately measure the outcome; for instance, symptoms can be variable, and ocular surface staining is open to observer bias. Use of minimally invasive objective metrics that respond to the treatment would enhance protocol design and interpretation of the results [132].
7.4 The role of symptoms in dry eye
Symptoms are, by definition, part of dry eye disease and in a recent review by the ODISSEY European Consensus Group members it was pointed out that for the majority of DED patients there is some relationship between symptoms and clinical signs, at least in severe disease [133]. However, it is also well established that perceived symptom severity may not equate to clinical signs of disease and that there exists a significant proportion of patients who have seemingly conflicting signs and symptoms. Part of the problem might be that symptoms are subject to significant variation over time, even within the same day [51,134] and that age- and sex-related, cultural, and ethnic influences on symptoms have to be taken into consideration as well [135,136].
To value whether the change of a clinical sign or symptom in dry eye disease is to be considered meaningful or important can be very challenging, especially given the limited correlation between signs and symptoms [80, 137]. In a study by Miller et al., the MCID for the Ocular Surface Disease Index (OSDI; Allergan Inc., Irvine, California) was assessed to better evaluate whether a statistically significant change in the OSDI score due to a given therapy in a clinical trial actually matters to the patient using an anchor based method. The results revealed an MCID ranging from 4.5 to 7.3 for mild or moderate disease and from 7.3 to 13.4 for severe disease [138]. Sullivan et al. reported that up to 40% of subjects with clear objective evidence of DED do not report symptoms on the OSDI. When symptoms are present, they can be useful, but their absence does not rule out the presence of disease [51].
Several different patient-reported outcome questionnaires, such as the 25-item National Eye Institute Vision Functioning Questionnaire (NEI-VFQ) [139], Dry Eye Questionnaire (DEQ) [140], Impact of Dry Eye on Everyday Life questionnaire (IDEEL) [141], the Ocular Surface Disease Index (OSDI) [142], and the Symptom Assessment in Dry Eye (SANDE) [143], have been developed to identify dry eye disease and to monitor changes in patients symptoms and life quality during a clinical trial. The OSDI and SANDE have been shown to correlate well [67]. It should be noted that the OSDI is copyrighted by Allergan, Inc. and the SANDE is copyrighted by Schepens Eye Research Institute, but both can be licensed for use in clinical trials. Furthermore, symptoms can also be graded using a visual analog scale (VAS) that could measure the most common symptoms of DED or be focused on the most important symptom identified by each patient.
The FDA suggest that patient-reported outcomes questionnaires should be based on a clear conceptual framework and that there should be evidence supporting their psychometric properties. Furthermore, the FDA also recommends specification of the MCID as a benchmark for interpreting the mean score differences between treatment arms in a clinical trial. (Guidance for Industry: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims, December 2009; http://www.fda.gov/downloads/Drugs/.../Guidances/UCM193282.pdf).
7.5 New functional outcome measures
Patients with DED often present with complaints of reduced visual function [144]. These manifest as symptoms of glare and blurred or fogged vision, which can adversely affect quality of life [142,145]. Snellen or logMAR charts (e.g., the Early Treatment Diabetic Retinopathy Study [ETDRS] chart) are typically used to measure visual acuity (VA). Since the reduction in visual acuity seen by DED patients is usually transitory and can be overcome by increased blinking, it is difficult to show differences in visual function between patients with DED and those without the disease with this method. The same is true for clinical trials that attempt to evaluate treatment effects via changes in VA.
A way to detect changes in vision in DED patients is to measure functional VA, with a method such as spatial-contrast sensitivity [146,147]. Rolando et al. found that spatial-contrast sensitivity was significantly lower in patients with DED compared with a group of age-matched normals. Contrast sensitivity has been shown to improve in dry eyes after the administration of artificial tears [148]. Functional VA can be studied also in terms of reading speed. Ridder and colleagues [149] demonstrated that reading performance decreases with increasing severity of dry eye and that this measure might even be used to monitor improvement with treatment in patients with DED.
Other functional visual tests have been used to identify deficits in the dry eye patient population. Goto and colleagues found that functional VA reduced significantly in patients with DED after gazing for 10–20 s without blinking. In addition, blinking rate during reading and driving were significantly reduced [150]. Another study used a continuous functional VA measurement system to evaluate monocular recognition acuity during a 30-s period of no blinking [151]. Functional VA in patients with dry eye was significantly lower than those of control subjects at all time points.
Another method to measure functional VA is to study higher order aberrations in patients with DED [152]. Dry eye patients with superficial punctate keratitis experienced significant deteriorations of visual function and optical quality compared with dry eye patients without staining and normals, as measured by the variation of coma-like and total higher order aberrations. These results support the hypothesis that optical disturbances from DED in the central portion of the cornea can affect functional visual performance. The progressive degradation of ocular optical quality has been shown to result from the loss of contrast at intermediate and high spatial frequencies in DED patients. The same study found that the progression index for corneal higher-order aberrations was correlated with objective clinical findings of tear film and ocular surface damage and the subjective index of patient-reported visual outcomes. There are encouraging signs that these are reliable measures of visual function. The TFOS DEWS II Diagnostic Methodology report includes additional information [56].
7.6 Biomarkers and surrogates
“A biomarker is a characteristic that is measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacological responses to a therapeutic intervention and a surrogate endpoint is a biomarker that is intended to substitute for a clinical endpoint and is expected to predict clinical benefit (or harm or lack of benefit or harm) based on epidemiologic, therapeutic, pathophysiologic, or other scientific evidence.” [153] Other terms used for this concept are endpoints and outcomes.
Some of the most frequently used biomarkers in DED clinical trials are ocular surface staining, tear protein levels such as inflammatory markers, or a physical property of the tears (e.g., osmolarity, lipid layer thickness). Some of these measures may be altered in patients with DED [154]. While there is a lack of correlation in most patients between the symptoms and these biomarkers until a substantial level of disease severity is achieved (i.e. moderate to severe disease), their overall value is substantial [155,156]. It should be noted that at the present time no specific biomarker has been successfully used in FDA registration trials for treatments for DED.
In an attempt to better define DED, the development of a composite score of biomarkers has been proposed. The methodologies employed include: independent component analysis (ICA), and latent class analysis (LCA). The former objective approach, using 7 equally weighted variables, revealed a number of clinically significant findings, including that many subjects with positive objective tests do not report clinically significant symptoms, and that corneal staining is a relatively late manifestation of disease. Using this composite score, only one variable biomarker, tear osmolarity, was found to parallel severity as measured in the composite scale across the entire spectrum of severity [129]. In another study comparing the results of ICA with LCA, tear osmolarity was found to be the best single marker of disease with ICA but not with LCA [128]. Note that the LCA method is dependent upon a number of assumptions and involves complex mathematical maneuvers. On the other hand, ICA can be readily utilized in a clinical setting.
A number of inflammatory cytokines have been measured in patients with DED [157–159]. The change in biomarkers with effective treatments is a current area of research. The utility of a new biomarker or surrogate must be evaluated on the basis of its sensitivity and specificity. The use of biomarkers in clinical trials throughout medicine is an area of active interest (FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource; http://www.ncbi.nlm.nih.gov/books/NBK326791/). Additional information may be found in the TFOS DEWS II Tear Film report [131].
7.7 Recommendations for regulatory review of clinical efficacy
The previous comments in this review emphasize that DED is a complex clinical condition for which to conduct clinical trials, particularly when assessing the effect of novel treatments in RCTs. This is due, in part, to the need for a definition that is clear, comprehensive, and encompassing of the current understanding of the disease characteristics. It is critical that the classification of a multi-factorial disease include the major subtypes of the disorder (e.g., aqueous tear deficiency and evaporative dry eye), while recognizing that there is a large contingent of mixed mechanism of disease. Also important is the proper determination of severity of disease with criteria for assessing severity both by categorical and continuous methodologies [129]. The recognition of the role of instability of the tear film that is observed in subjects with dry eye must be contrasted with the stability present in normal subjects. Similarly, the bilaterality of DED with its effects on both eyes should be identified with recognition of the subsequent negative effect of a priori choice of a treatment eye. Finally, there must be adequate controls, allowing for placebo-nocebo effects in clinical trials with recognition of the lubricant and potential tear stabilization effect of many agents used as controls [59]. The lack of approval for registration of new therapies for over a decade suggests that adjustment of the design and outcomes assessment of clinical trials in treatment of dry eye disease is required while observing the tenets of good research practice [38,160].
7.7.1 Symptom versus sign in outcome parameters
The disparity between symptoms and signs in DED presents a severe challenge to achieving approval if an effect on both signs and symptoms is required in the same trial [122]. The FDA acknowledges this dilemma. In its 2016 approval of Xiidra® (lifitegrast), it set a precedent of allowing separate trials to demonstrate efficacy in a sign and a symptom. This represents one possible solution to the difficulty. Improved selection of subjects entered into clinical trials with attention to variability of pain thresholds or level of ocular surface sensitivity should be considered. The effects of tear instability or disruption of tear homeostasis [2], a hallmark of DED, on all of the commonly used endpoints in subjects with DED should be recognized. The determination of variability in symptoms must account for the effects of neural impairment of the ocular surface, which has been demonstrated to occur in DED including hyperalgesia in early disease and hypesthesia in advanced disease [161,162]. The entity of keratoneuralgia can be a serious confounder in clinical trials, and correction of symptom level with respect to the above is needed [163]. Likewise, the variability of levels of physical and visual activities of enrolled subjects during the clinical trial can complicate demonstration of effect. Therefore, a more precise correlation of improvement in a symptom with level of activity or environmental exposure during a trial may be required.
7.7.2 Subjective versus objective outcomes
Some features of DED may require assessment of functional outcomes, since visual disturbances are symptomatic to the patient and reduce quality of life but have generally not been included in previous therapeutic clinical trials [144]. Analysis of functional visual ability allows objective measure of visual impairment and can be accomplished by assessing the ability to read or engage a video display terminal [149]. A specific test, named “Functional Visual Acuity”, can be measured at relatively short intervals [164]. Impaired contrast sensitivity may also be a monitor of the adverse effect of DED on visual performance [146]. Lastly, clinical correlation with analyte measurement of a biomarker could serve as an objective outcome in clinical trials in DED.
7.7.3 Appreciation of biomarkers
Many new biomarkers of DED have been identified; some have been studied extensively as possible indicators of DED (e.g., osmolarity and inflammatory proteins) [154,165]. The correlation of biomarkers with symptoms may require an approach as identified in Section 7.7.1.
7.7.4 Approaches to clinical trial design
Changes to the approach to clinical trial design often entail additional cost and complexity. This is probably why some of the recommendations made in the 2007 DEWS report have not been implemented, but such changes will be needed to allow qualifying treatments for registration. Regulatory agencies will need to be receptive to such alternative proposed designs. Some considerations for future clinical trial design should include withdrawal trials, in which all subjects entered receive the active treatment while a randomized number switch to placebo/vehicle control. This has been used in other disease states with some success, but requires that the active therapy improve but not cure the DED [8–12]. Another option is to identify biomarker outcomes with rigid control of measurement protocols at all investigative sites and with appropriate calibration of instrumentation.
It would be important to include both eyes in the trial since each eye is inter-dependent and differences between eyes document the dynamic range of the tear instability present. This reflects the random distribution of effects of breakdown in the tear-ocular surface homeostasis in DED. Failure to do so will inevitably lead to loss of valuable information [52]. However, appropriate methods must be used to count only one statistic per patient.
In an effort to decrease variability and standardize clinical trials of DED therapy, and in the same way that a controlled allergen challenge model has been successful in evaluating and developing novel treatment for allergic conjunctivitis [166], a controlled adverse environment (CAE) has been evaluated for treatments for DED [167,168]. The CAE has been used to both screen subjects and to evaluate on-treatment effects [169–171]. However, treatment effects typically have not been seen in a priori outcome measures, but rather in secondary measures, of which there are many. As well, to date, there are few publications of Phase 3 clinical trials using the CAE, nor, like some environmental studies, publications confirming replication of results. The role of the CAE in drug development vis-à-vis clinical environmental studies is not clearly understood at this time.
8.1 Preclinical safety to support clinical studies
This report focuses on the design and conduct of clinical studies to evaluate therapeutics for DED, particularly novel therapeutics. An important question in the development of new products is the requirement for nonclinical safety studies to support the intended clinical evaluation. This is covered in ICH M3 (R2) (“Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf). As an example, Table 3 of this guideline (“Recommended Nonclinical Studies to Support Exploratory Clinical Trials'’) illustrates how the required timing and amount of nonclinical studies depend very much on the perceived hazard. In general, the safety studies required are driven by the clinical studies planned. For example, a clinical study of one week duration requires nonclinical toxicology studies of at least one week duration using a dose exaggerated as to the anticipated clinical dose. Note that regulatory authorities do not require proof of efficacy in animal models for drugs or biologics [172]. One should have an idea as to what administered dose would provide a therapeutic concentration at the target site. The ICH M3 (R2) guidance is relatively limited on ophthalmic drugs. For local ocular dosing, an ideal treatment should be local, that is, an ideal local treatment does not result in meaningful systemic exposure. The systemic exposure after ocular dosing is key to determining the level of systemic toxicology studies that need to be conducted [173]. Issues surrounding nonclinical safety plans for ocular drugs and biologics is discussed in Novack and Moyer [174]. Medical devices require biocompatibility testing.
8.2 Safety assessment during clinical studies
As is the case with all clinical decisions, the decision to enroll a subject in a clinical trial balances the benefits to the subject with the risks of being in the trial. One of the fundamental purposes of the clinical development program is to understand the safety profile of the drug and not to put subjects at unnecessary risk. The collection of safety information is an ongoing process, and more data are collected as the drug moves through clinical development. Early on, the safety profile is based on experience with compounds that have a similar mechanism of action to the drug being tested or a similar structure. This profile is supported by the pre-clinical program and clinical pharmacology and is updated as new information is collected. The most up-to-date safety information is described in the investigator's brochure (IB). This should be carefully read by all investigators prior to enrolling subjects in a clinical trial. Ultimately, the safety information collected during the clinical development program is described in the package insert. Additional safety information is updated as post-marketing experience warrants. The collection of safety information about a drug continues throughout a drug's lifetime. Many of the issues regarding clinical safety are covered in ICH E6. While the focus of products administered locally to the eye is ocular, and the principal investigators are typically eye care professionals, nonetheless there is a requirement to evaluate systemic safety as well.
8.3 Phase 1 clinical trials
Phase 1 trials are designed to evaluate safety and tolerability. They may be conducted in healthy volunteers or in patients with the disease of interest depending on the risk-benefit profile of the drug. Phase 1 is typically when pharmacokinetic data is first assessed, and subjects may be confined for a short period of time to a phase one unit for close monitoring. Dose escalation to find the maximum tolerated dose is typically done during phase 1 trials. Ocular and systemic evaluations are performed based on potential toxicities of the compound being evaluated. Vital signs, systemic chemistry, and hematology are commonly evaluated. For biologics, immunogenicity is typically assessed at early time points after drug exposure and later time points after exposure.
Although the risks of systemic toxicity from topical ocular dosing are less than the risks from oral dosing, the fractional absorption following topical ocular administration is usually about 0.9. In other words, an eye drop has about 90% of the systemic availability of an intravenous injection of the same amount of drug [173]. This systemic availability is partially dependent upon the drainage of the tears through the nasolacrimal duct into the vascularized nasal and pharyngeal region, as well as conjunctival vessels. Drug absorbed in this manner may be delivered to the heart without the potential for hepatic metabolism. Because the total dose contained in an eye drop is usually much less than that administered intravenously or orally, the systemic safety margin is usually large. That said, potent drugs such as β-adrenoceptor antagonists and α2-adrenoceptor agonists may be absorbed in amounts significant enough to cause negative cardiovascular effects [175]. Furthermore, idiosyncratic reactions to biologics (e.g., the “cytokine storm” provoked by TG1412, a superagonistic anti-human CD28 antibody, that caused multi-organ failure in human volunteers exposed to 1/500th of the no-observed-adverse effect level in primates) also are worth taking into consideration in considering dose selection for first-in-human studies.
8.4 Phase 2 and phase 3 clinical trials
As a potential therapeutic is developed through later stage trials, the duration of treatment, subject population, and dosing regimen increases, approaching that of the anticipated therapeutic use. Safety is still of primary concern, as the number of subjects exposed is still relatively low.
8.5 Adverse events
Adverse events are any untoward medical occurrence associated with the use of a drug in humans whether or not considered drug related (21 CFR 314.80). Adverse events may be a specific disease, sign, symptom, or abnormal laboratory value or imaging study. For subjects with pre-existing conditions or laboratory values, an adverse event may represent a worsening of the condition or value while in the study. Adverse events may be graded on severity from mild to life threatening.
8.5.1 Serious adverse events
Serious adverse events (21 CFR 313.32(a)) are any adverse events that in the judgement of the investigator or sponsor results in:
1. Death or immediate risk of death,
2. Inpatient hospitalization or prolongation of existing hospitalization,
3. Persistent or significant incapacity or substantial disruption of the ability conduct normal life functions or
4. Congenital anomaly or birth defect. All serious adverse events must be reported by the investigator to the sponsor regardless of causality with appropriate supporting documentation.
8.5.2 Unexpected adverse event
An unexpected adverse event (21 CFR 312.32(a)) is one that is:
1. Not listed in the Investigator's Brochure,
2. Not listed in the Investigator's Brochure at the specificity or severity observed, or
3. Mentioned in the Investigator's Brochure as anticipated due to the pharmacokinetic properties of the drug or occurred with other drugs in this class, but not with the study drug.
8.5.3 Serious unexpected Suspected Adverse Reactions (SUSAR)
Adverse events that meet these three criteria are SUSAR:
1. Serious (S),
2. Unexpected (U), and
3. Suspected Adverse Reactions (SAR).
SUSAR are reported to FDA or equivalent agency (and the IRB) through new expedited safety reporting rules (21 CFR 313.32(c)(1)(i).
8.6 Investigator's responsibilities
Clinical investigators are critical in ensuring high quality clinical studies. Investigators are responsible for following good clinical practice (GCP), which is different than following good clinical practice when treating and evaluating patients outside of clinical trials. GCP ensures the quality and integrity of the data collected and ensures that the rights, safety, and welfare of subjects are protected. Investigators must personally conduct the study, ensure all persons assisting in study are informed of obligations, follow the protocol, ensure informed consent of study subjects, keep appropriate records, properly account for study drug and comply with other requirements in (21 CFR 312.32 and 21 CFR 314.80).
9. Benefit/risk assessment back to top
Key to any therapeutic intervention in a patient is an evaluation of benefit vs. risk (benefit/risk). This is based upon the premise that any intervention carries with it some risk. In classic pharmacology, one determines the dose-response for both the desired efficacy, and for the undesired toxicity or toxicities. The ratio between the toxic and effective doses is called the therapeutic index. As noted above, in the US, the passage of the Kefauver-Harris amendments in 1962 required that efficacy be part of the approval of new drugs. As safety was already required (Federal Food, Drug and Cosmetic Act as passed in 1938), this now gave the FDA the regulatory basis to judge efficacy in light of safety, i.e., benefit/risk for therapeutics [176]. There are no published guidelines on exact ratios allowed, rather it is a decision made on a drug-by-drug basis (and indeed, a device-by-device basis). However, when considering the limits of bioequivalence for approval of generic drugs, regulatory agencies may judge agents with narrow therapeutic indices and high pharmacokinetic variability more carefully than others (e.g. warfarin, levothyroxine, carbamazepine, and phenytoin) [177,178].
The current director of the FDA's Center for Drug Evaluation and Research, has started discussions stating “a common framework is necessary for the transparent articulation of the benefits and risks of a therapeutic product across disparate stakeholders.” Proposals might include a restricted approval for a limited patient population, with commitments for larger, longer studies for wider approvals [179]. Academic and industrial researchers are working on quantitative methods for assessing benefit/risk. This benefit/risk ratio is used in drug approvals, formulary decisions on the optimal therapeutic agent, and corporate decisions on selection of which molecule to develop [180–183].
The issue of benefit/risk ratio for an ophthalmic drug was in the news in 2012 with the non-approval of Alimera's Iluvien® (fluocinolone acetonide intravitreal implant) in the U.S. for the treatment of diabetic macular edema (DME). While there had been a clear demonstration of efficacy with the both phase 3 trials successfully meeting their primary endpoint, the FDA had judged that the benefit did not outweigh the risks in DME [184]. In Europe, this concern was addressed through a pre-planned subgroup analysis, where a larger treatment effect was seen in the patients with chronic DME, and the product was approved in 2012. In 2014, FDA addressed their benefit/risk concern by requiring that only DME patients who have received a prior course of corticosteroid and did not demonstrate a clinically significant rise in IOP, would be indicated for treatment with Iluvien®. The product was subsequently approved in the U.S. for this subset of patients where there was demonstrated improvement in the benefit/risk ratio.
With respect to treatment of dry eye, many of the new chemical entities evaluated in the pharmacotherapy of dry eye have little or no ocular or systemic safety issues. Thus, any evaluation of benefit/risk ratio mathematically solves to efficacy only. Corticosteroids, long used to treat a myriad of ocular inflammatory conditions [185], are also known to treat dry eye [69]. However, clinicians know that chronic use of corticosteroids, irrespective of potency, have the risk of elevating intraocular pressure and causing cataracts in phakic patients. Thus, their utility in treating dry eye is at present, a case of application of therapeutic index.
Regulatory requirements in dry eye disease vary across different territories worldwide. Sponsors conducting clinical trials in one territory often find these trials are not sufficient to support approval in another territory. Differences in clinical trials across various territories include: endpoints, comparator, need to show clinically meaningful difference between active drug or device and comparator, inclusion of quality of life metrics, demonstration of mechanism of action, and length of trial. The worldwide regulatory situation for DED therapies has been reviewed [7,39].
10.1 Drugs
10.1.1 United States regulatory requirements
Regulatory approval in the US requires that the sponsor demonstrates a statistically significant benefit of the active drug compared with the vehicle in two adequate and well controlled trials. The benefit of the active drug needs to be demonstrated for both a sign of the disease (for example, corneal fluorescein staining) and a symptom of the disease (for example, feeling of dry eye) in both trials at pre-specified time points. There is currently no regulatory requirement for a clinically meaningful difference between the active drug and comparator. For approval of a new drug application (NDA) or biological license application (BLA), the FDA will consider either negative-control (where superiority is required) or positive-control studies (where the investigational product must be at least as effective as the control). For the latter, the positive control product must be approved in the US or superiority is required. While not needed for regulatory approval in the US, clinical relevance may be important in the United States for payers. The FDA does not mandate duration of the trial for efficacy, but for therapy for DED, requires that at least 100 subjects be exposed to the intended dose of the drug or higher chronically, which is typically 6–12 months.
As noted elsewhere in this document (See Section 5.1), worldwide, approvals for treatments for treatment of DED are relatively few. Two pharmacological products are approved for treatment of DED in the US (as of July, 2016), Restasis® (cyclosporine ophthalmic emulsion) is technically approved to “increase tear production in patients whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca.” (Restasis® package insert). The other topical medication is lifitegrast (Xiidra®), an ophthalmic solution approved to treat the signs and symptoms of dry eye disease. Lifitegrast is an antagonist of LFA-1 thought to block signaling of inflammation in DED [186].
As an additional consideration, the TFOS DEWS II report has highlighted DED in populations with limited focus previously, such as DED in younger age groups, as information on new risk factors emerges [42]. This may have implications for the generalizability of clinical trial results and may be a key consideration in approval of new pharmaceutical products. For trials in younger patient populations in the US and EU (and perhaps elsewhere), a Pediatric Study Plan (US)/Pediatric Investigation Plan (Europe) is required when a product goes to Phase 3. The natural history of the disease in pediatric patients is key for such plans.
The safety and efficacy of studies on autologous tears are covered in the TFOS DEWS II Management and Therapy report [14]. In this report, we present the regulatory perspective on this therapy. The US FDA recently issued a guidance on the homologous use of human cells, tissues, and cellular and tissue-based products (HCT/P). (http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM469751.pdf),
This guidance makes reference to 21 CFR 1271, specifically 21 CFR 1271.10(a)(2). This guidance in turn suggests that if HCT/P does not involve the combination of the cells or tissues with another article, except for water, then pre-market review by FDA is not required (i.e., an NDA or Biologics License Application (BLA) submission). However, with respect to preparing autologous tears for patients, which requires an outside sterile laboratory for preparation sometimes in another state, those facilities are governed by Section 503B of the Federal Food, Drug, and Cosmetic Act, under the heading Registered Outsourcing Facilities. These facilities are in turn governed under a relatively new guidance on outsourcing facilities (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM434171.pdf). The facilities must comply with cGMP. As well, a short shelf life for the product, even refrigerated, is required, in the order of 5 days. This has effectively limited the use of autologous tears in most of the. Note that these same issues are challenging the availability of liquid lissamine green solution. We also sought information on the regulatory position in other countries (e.g., Agência Nacional de Vigilância Sanitária (ANVISA) in Brazil, etc.) but were not able to obtain clear direction at the time of writing. Jones et al. [14] suggest that one problem in evaluating this therapy is that there is no consistent method for the preparation of autologous tears across laboratories. While by definition this is personalized therapy, meaning it is the patient's own blood used to derive the therapy, nonetheless, a move towards standardization of the preparation in order to clearly evaluate it in a controlled study, might be moot in the current regulatory environment.
10.1.2 European regulatory requirements
In contrast to the US, pricing is integral to the approval process in Europe. Thus, if there is an approved product in EU, the pivotal clinical trials should be conducted as positive-controlled studies against an approved product. Again, with the caveat of few approved examples, European perspective appears to be that the benefit of the new over existing products does not need to be shown for both a sign and symptom of the disease and that approval could be based on showing a benefit for a sign or a symptom of the disease. As with the US, European authorities encourage Sponsors to include quality of life metrics. In our experience, European authorities may expect more clinical information on the mechanism of action, and with a range of patients with DED, and for longer periods. As noted previously (Section 7.2), in March 2015, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) granted marketing authorization for Ikervis® (cyclosporine emulsion 0.1%), based on improvement in corneal damage and demonstration of anti-inflammatory mechanism of action in the most severe dry eye patients. The drug did not show a benefit over vehicle in treating the symptoms of dry eye disease [187].
10.1.3 Japanese regulatory requirements
There are several agents approved in Japan to treat DED that are not available in North America or Europe [7]. These include: sodium hyaluronate (Hyalein® 0.3), approved in 1995, with a primary mechanism of increasing tear film stability; diquafasol tetrasodium (Diquas® Ophthalmic Solution 3.0%), a P2Y2 receptor agonist approved in 2010 and rebamipide (Mucosta® Ophthalmic Solution 2%) approved in 2011, which are marketed as secretagogues [7,188,189]. Also available in Japan are a cyclosporine product (Papilock™ mini-ophthalmic solution 0.1%), and a tacrolimus product (Talymus™ ophthalmic suspension 0.1%), both approved for the treatment of vernal keratoconjunctivitis.
10.2 Devices
In the US, medical devices are regulated by the FDA's Center for Device and Radiological Health (CDRH). For medical devices, the FDA uses established risk-based classification criteria to classify a device as Class I, II or III. The established risk-based criteria are determined, in part, from what is already known regarding comparable existing devices (also known as “predicate devices”). Class I devices are subject to general agency controls (such as accurate branding, submission of device records and reports, quality system regulation, and so forth). The LipiView® (TearScience, Morrisville, NC), a device to image the lipid layer of the tear film and meibomian glands, is an example of a Class I device [190,191]. In this case, the agency considered the device to be substantially equivalent to that of digital ocular photography. For Class I devices, these general controls are considered sufficient to provide the agency with reasonable assurance of the device safety and effectiveness and often a 510(k) process is not required. The majority of Class I devices are exempt from trial requirements (e.g., manual instruments, bandages, casting).
For class II devices, general controls alone are not sufficient and they require specific agency controls such as RCTs, guidelines for the submission of clinical data, animal testing, standards, post market surveillance and patient registries. Typically, a 510(k) process is required. The agency considers the general and specific controls sufficient to provide reasonable assurance of safety and effectiveness of the device. The LipiFlow® (TearScience, Morrisville, NC), is an example considered by the agency to be a Class II device. The LipiFlow was subject to a 510(k) de novo process and was cleared for the application of localized heat and pressure therapy in adult patients with chronic cystic conditions of the eyelids, including MGD [107, 116]. (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/denovo.cfm?ID=DEN100017) [112,192]. A similar regulatory route was also employed for the recently cleared TrueTear™ (intranasal neurostimulating device, Allergan) [193].
The FDA will classify a device as Class III if general controls are not adequate to provide reasonable assurance of safety and effectiveness and there is not sufficient information to establish special controls that would provide such assurance. This is typical of devices for which there is no reasonable predicate device, or if the device presents substantial risk to the consumer. Class III devices are required to go through an extensive pre-market approval (PMA) process, whereby all of the necessary scientific and clinical information is gathered for the FDA in order to provide reasonable assurance of safety and effectiveness of the device [194]. These include life sustaining or life supporting or novel technology, such as pacemakers or respirators.
The device approval process in the USA typically follows one of two steps:
1. A premarket notification (PMN) or 510(k) submission and clearance is required in order to market the product. Typically, a predicate device (previously cleared device similar to the new device) is referenced to demonstrate that the new device is as least as safe and effective as (substantially equivalent to) the predicate device. http://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketnotification510k/ucm134572.htm
2. PMA is a much more stringent device application. The PMA application contains information about how the medical device was designed and how it is manufactured, as well as preclinical and clinical studies of the device, demonstrating that it is safe and effective for its intended use. The PMA route requires a clinical trial and thus is a much more lengthy and expensive process than a 510(k) application. http://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketapprovalpma/default.htm
To clarify the distinction between the PMA and 510(k) designations. PMA indicates “approval” based on safety and efficacy for a designated indication, whereas a 510K indicates “clearance” that a device is safe and does not comment on its efficacy.
While regulatory information usually provides information on safety, efficacy may not be addressed for devices upon approval, in which case post-regulatory approval trials need to address this issue. Such clinical trials may or may not be undertaken and may not provide sufficient information for absolute determination of its efficacy.
Surgical interventions are typically not under regulatory review unless they include a new device or new pharmaceutical that is not otherwise approved. In these cases, determination of efficacy and safety is based on available information that may or may not be peer reviewed and may or may not use standardized clinical trial designs. For instance, Geerling describes the transplantation of the major salivary glands to treat DED with both a case series [195] and a literature review [196].
A key resource for statistical planning, analysis and reporting of clinical trials is the ICH E9, “Statistical Principles For Clinical Trials” [197]. This guidance presents topics relevant to clinical trials in dry eye including: population, primary and secondary variables, composition variables, techniques to avoid bias, pros and cons on different designs (e.g., parallel vs. crossover), consideration for multicenter trials, and superiority, non-inferiority, and equivalence. A key feature of this guidance is the a priori selection of outcome measures, the penalties for interim analyses, and post hoc analyses (i.e., moving the target after the arrow is shot) [198].
Of particular import in trials of dry eye therapies is the handling of efficacy endpoints of signs and symptoms. The issue of multiplicity is discussed in Section 2.2.5 of ICH E9, which states: “If the purpose of the trial is to demonstrate effects on all of the designated primary variables, then there is no need for adjustment of the type I error, but the impact on type II error and sample size should be carefully considered.” Stated differently, if you require both the sign and the symptom to declare success, then you can test both at p ≤ 0.05. If you require only one to be successful, then you must test both at p ≤ 0.025. Another alternative is to declare one endpoint to be first in a hierarchy of endpoints and plan to test them in a pre-specified order (the hierarchy). You can declare each as statistically significant at p ≤ 0.05 until you reach one that is not, and then you stop. Interpretation of such an outcome is open to discussion, but typically this would NOT be adequate for regulatory approval.
Calculation of power for a co-primary efficacy endpoint is more complicated than for a single efficacy endpoint. One needs to take into account the correlation between the two endpoints, the more highly correlated the greater the reduction in sample size that is needed. For dry eye the two endpoints are typically poorly correlated, and so one can typically ignore this potential benefit on sample size [199], and use the endpoint with the lower effect size for calculations.
An example of a co-primary endpoint used in a large controlled phase 3 trial is a study of lifitegrast [62]. As noted previously (Section 7.2), the SANSIKA trial of Ikervis® (cyclosporine emulsion) initially planned a primary outcome of signs and symptoms. The trial was unsuccessful in meeting this outcome, but a subsequent analysis of improvement in corneal fluorescein staining showed a statistically significant difference in favor of Ikervis®) [130,187].
Simply stated, an investigational active treatment must be superior to an inactive treatment in order to be considered effective (superiority study). An alternative approach is to show that the investigational active treatment is equal to another active treatment. The concept of equality is considered frequently in statistical tests of trials of comparative therapeutics. More recently, the concept of “non-inferiority” [200], has gained prominence. This is covered in guidances from the European Medicines Agency (EMA) (2002), ICH E10 (2001) and FDA (2010). Four key criteria are noted that must be met in a non-inferiority trial: 1) the active control must be effective; 2) there must be an acceptable non-inferiority margin, 3) the design of the comparative trial of the new treatment must be similar to that of the original studies; and 4) the conduct of the trial must be of high quality. The non-inferiority margin is a difference that is small enough to be considered clinically insignificant. Examples of possible outcomes of trials are shown in Fig. 2 [201].
Fig. 2 Three possible results of a placebo-controlled superiority study (point estimate, 95% confidence intervals).
Assume that the response variable is positive and a larger value indicates a better response.
1. Point estimate of effect is 2; 95% CI lower bound is 1. Conclusion: Drug is effective and appears to have an effect of at least 1.
2. Point estimate of effect is 2; 95% CI lower bound is < 0 (study perhaps too small). Conclusion: Drug is not shown to be effective.
3. Point estimate of effect is 0; 95% CI lower bound is well below 0. Conclusion: Drug shows no suggestion of effectiveness.
C = Control; T = Test, M = mean, NI = non-inferiority, CI = confidence interval.
From: Guidance for Industry: Non-inferiority clinical trials. U.S. FDA, March 2010 (Reprinted from Novack GD: Some are more equal than others. Ocul Surf. 2014; 12:155–8.)
For either a superiority or non-inferiority study, GCP and ethics state that the sample size must have adequate power to detect the difference. The formula for power includes five variables: power (also known as beta, the chance to find a difference if it exists), the clinically significant difference (or the non-inferiority margin), the sample size, the variability, and the alpha level. The smaller the difference to be detected, the larger the sample size required [202]. Power can be calculated using various statistical programs (e.g., SAS, Nquery), as well as websites (e.g., http://powerandsamplesize.com/). The size of a clinical study depends upon the phase of development (See Section 4.2), the sample size required for efficacy, and the sample size required for safety (See Section 4.2), as well as adjustments for disqualified and discontinued subjects. Diagnostic methods to evaluate DED are provided in the TFOS DEWS II Diagnostic Methodology report [56]. For example, if a researcher wanted to have 80% power to detect a clinically significant difference of at least 0.5 units (0–3 scale) in corneal staining, which had a standard deviation of 1 unit, a sample size of 64 subjects per group would be required. Adjusting for disqualifications and discontinuations of 20%, then ∼77 subjects per group would be required. However, if this was one of two Phase 3 trials, as part of a development program, it would be part of a the minimum safety requirements of 300–500 patients on the drug (any dose) and 100 at the intended dose or higher, then the sample size per group might be increased to 180 per group in order to obtain an adequate sample size.
12. Recommendations for future directions in dry eye clinical trials back to top
Review of past peer-reviewed publications on drugs and biologics for DED treatment points to the difficulty of evaluating efficacy and safety. Many of these studies do not meet the current recommendations of this group for maximizing quality and interpretation for pivotal trials [61,62,69,70,76,88,93,127,188,203–208]. In order to improve the quality of clinical trials going forward, to optimize resources, and increase the opportunity for novel therapeutics for patients with DED, the TFOS DEWS II Clinical Trials subcommittee has the following recommendations.
First, that studies be conducted consistent with Good Clinical Practice (GCP). This involves using GMP-quality clinical trial material. While this may be a daunting task, clinical trialists should consult colleagues and drug development experts who are familiar with this system of controls. This includes appropriate protections for the study subjects. GCP also requires compliance with appropriate regulatory requirements in the jurisdiction of study conduct, and may require additional regulatory filings if the investigational shipment is prepared and shipped from another state or country. While the use of the CONSORT statement is of limited value at the end of study when a manuscript is being prepared, it is useful to review prior to planning and starting a study.
Next, the subcommittee recommend that the design, treatments, and sample size be consistent with the investigational treatment, the objectives of the study, and the phase of development. For example, a crossover or paired-comparison design may be appropriate for a comfort study in normal volunteers, but not so for a long-lasting treatment with the potential for systemic or contralateral effects. Also, the dose of a drug or biologic should not only be less than that which was toxic or not tolerated in nonclinical or previous clinical studies, but also at a dose and frequency to provide therapeutic concentrations at the intended site of action. The duration of treatment, at least for a pivotal study, should also be consistent with the mechanism of action and time-course of effect.
For pivotal studies, sample size is key to the potential validity of the study. An underpowered positive-controlled study will find apparent equivalence, which may be the incorrect conclusion of an appropriately powered study.
Outcome measures are key to determine the efficacy of the treatment and, if possible, should include minimally invasive objective metrics that are in line with the expected mechanism of action of the treatment. Exploration of new ways to evaluate dry eye disease, such as biomarkers, may lead to improvement in dry eye trial design and increased clarity on the efficacy of a new treatment.
The range of new devices for diagnosing and treating dry eye disease is expanding. The use of these new devices in diagnosing and selecting treatment for patients with DED, as well as their use in evaluating novel therapies will require clinical data that exceeds the typical regulatory requirements for device approval (which tend to focus on safety and device reliability). Note that measurement of dry eye signs and symptoms is covered elsewhere in the TFOS DEWS II Diagnostic Methodology report [56].
In this review of published clinical studies of therapeutics for DED, many reports are less than adequate in describing the methods and analysis. For example, the pharmaceutical make-up of the placebo, or the nature of masking is not clear. With the caveat that printed journal space may be limited, reporting should be as complete as possible so that the study may be appropriately evaluated. Some journals (e.g., Ophthalmology) provide for details in addition to the printed journal to be available in the on-line publication. This caveat is moot for regulatory submissions, as these requirements are part of GCP and ICH guidances.
13. Financial disclosures back to top
G. Novack: Acucela, Allysta, Bausch + Lomb, Eleven Biotherapeutics, Oculeve, Panoptica, Portage, Senju Pharmaceutical Co., Ltd., Shire (C).
P. Asbell: Alcon, RTech (F); Bausch + Lomb, Eleven Biotherapeutics, Nicox, Oculus (C); Alcon, RTech (R).
S. Barabino: Alcon, Farmigea, TRB Chemedica (R).
M. Bergamini: Nicox (purchased by Valeant) Aug 14- Oct 15; Aciex (wholly owned subsidiary of Nicox, S. A. Oct 14 - present (E); Nicox, S. A. XOMA US, LLC (C); Aciex Therapeutic, Inc., Nicox S.A., Nicox Inc. (R).
J. Ciolino: Aciex, Alcon, Bausch + Lomb, Celtic Therapeutic Development, EyeGate, Foresight Eye Therapies, Hi Tech, Jade Therapeutics, Johnson & Johnson, Merck, Mimetogen, Ocusoft, RegeneRx, Pfizer, SARcode, Seikagaku (SKK), Sun Pharma, (C); New England Cornea Transplant Research Fund, Foundation Grant, Mass Lions Eye Research Fund, Foundation Grant, Eleanor and Miles Shore 50th Anniversary Fellowships for Scholars in Medicine, Harvard Cornea Center of Excellence Fellowship, Research to Prevent Blindness, Career-Development Award (F).
G. N. Foulks: TearLab (I); Eleven Biotherapeutics, Inc; Healios, Inc; Insite Pharmaceuticals, Inc; Kala, Inc; Lexitas, Inc; Parion, Inc; R-Tech Ueno, Inc; and Shire Pharmaceuticals, Inc. (C).
M. Goldstein: None.
M. Lemp: TearLab (I); TearLab, TearScience, Santen, Europe (C).
S. Schrader: None.
C. Woods: None.
F. Stapleton: Alcon Laboratories, Allergan, CooperVision, Johnson & Johnson Vision Care, Stiltec (F); Nidek (C).
F (Financial Support, I (Personal Financial Interest), E (Employment), C (Consultant), P (Patent), R (Recipient), N (No Commercial Relationship), S (non-remunerative).
[1] Design and conduct of clinical trials: Report of the Clinical Trials Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf 2007;5:153–162.
[2] Craig JP, Nichols KK, Akpek EK, Caffery B, Dua HS, Joo CK, et al. TFOS DEWS II Definition and Classification report. Ocul Surf 2017;15:276–283.
[3] Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38(6):727–735.
[4] Felson David T, Anderson Jennifer J, Boers Maarten, Bombardier Claire, Chernoff Miriam, Fried Bruce, et al. The American college of rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. Arthritis Rheum 1993;36(6):729–740.
[5] Sullivan BD, Crews LA, Sönmez B, de la Paz MF, Comert E, Charoenrook V, et al. Clinical utility of objective tests for dry eye disease: variability over time and implications for clinical trials and disease management. Cornea 2012;31(9):1000–1008.
[6] Sullivan BD, Whitmer D, Nichols KK, Tomlinson A, Foulks GN, Geerling G, et al. An objective approach to dry eye disease severity. Invest Ophthalmol Vis Sci 2010;51(12):6125–6130.
[7] Sullivan DA, Hammitt KM, Schaumberg DA, Sullivan BD, Begley CG, Gjorstrup P, et al. Report of the TFOS/ARVO Symposium on global treatments for dry eye disease: an unmet need. Ocul Surf 2012;10(2):108–116.
[8] Pocock Stuart J, Gersh Bernard J. Do current clinical trials meet Society's needs? J Am Coll Cardiol 2014;64(15):1615–1628.
[9] van Herwaarden N, van der Maas A, Minten MJ, van den Hoogen FH, Kievit W, van Vollenhoven RF, et al. Disease activity guided dose reduction and withdrawal of adalimumab or etanercept compared with usual care in rheumatoid arthritis: open label, randomised controlled, non-inferiority trial. BMJ 2015;350:h1389.
[10] Schmidt PJ, Ben Dor R, Martinez PE, Guerrieri GM, Harsh VL, Thompson K, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry 2015;72(7):714–726.
[11] Bajard A, Chaubaud S, Cornu C, Castellan AC, Malik S, Kurbatova P, et al. An in silico approach helped to identify the best experimental design, population, and outcome for future randomized clinical trials. J Clin Epidemiol 2016;69:125–136.
[12] Hertel J, Locatelli F, Spasovski G, Dimkovic N, Wanner C. Randomized, double-blind, placebo-controlled, withdrawal study of colestilan after dose titration in chronic kidney disease dialysis patients with hyperphosphatemia. Nephron 2015;130(4):229–238.
[13] Anonymous. International conference on harmonisation; good clinical practice: consolidated guideline; notice of availability. Fed Reg 1997;62:25694.
[14] Jones L, Downie LE, Korb D, Benitez-del-Castillo JM, Dana R, Deng SX, et al. TFOS DEWS II Management and Therapy report. Ocul Surf 2017;15:575–628.
[15] Anonymous. Guidance for industry CGMP for phase 1 investigational drugs. 2008.
[16] Anonymous. Guidance for industry - INDs for phase 2 and phase 3 studies - chemistry, manufacturing, and controls information. 2003.
[17] ICoMJ. In: Recommendations for the conduct, reporting, editing, and publication of scholarly work in medical journals. 2015.
[18] Chalmers I. Proposal to outlaw the term “negative trial. BMJ 1985;290:1002.
[19] Brice A, Chalmers I. Medical journal editors and publication bias. BMJ 2013;347:f6170.
[20] DeAngelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Clinical Trial Registration: A Statement from the International Committee of Medical Journal. JAMA 2004;292:1363–1364.
[21] Zarin DA, Tse T, Williams RJ, Califf RM, Ide NC. The ClinicalTrials.gov results database–update and key issues. N Engl J Med 2011;364:852–860.
[22] Zarin DA, Tse T, Ross JS. Trial-results reporting and academic medical centers. N Engl J Med 2015;372(24):2371–2372.
[23] Anonymous. Clinical trials registration and results information submission. Fed Reg 2016;81:64982–65157.
[24] Contreras JL. NIH's genomic data sharing policy: timing and tradeoffs. Trends Genet 2015;31(2):55–57.
[25] Strom BL, Buyse M, Hughes J, Knoppers BM. Data sharing, year 1-access to data from industry-sponsored clinical trials. N Engl J Med 2014;371(22):2052–2054.
[26] Doshi P, Dickersin K, Healy D, Vedula SS, Jefferson T. Restoring invisible and abandoned trials: a call for people to publish the findings. BMJ 2013;346:f2865.
[27] Doshi P. No correction, no retraction, no apology, no comment: paroxetine trial reanalysis raises questions about institutional responsibility. BMJ 2015;351:h4629.
[28] Le Noury J, Nardo JM, Healy D, Jureidini J, Raven M, Tufanaru C, et al. Restoring Study 329: efficacy and harms of paroxetine and imipramine in treatment of major depression in adolescence. BMJ 2015;351:h4320.
[29] Hudson KL, Collins FS. Sharing and reporting the results of clinical trials. JAMA 2015;313:355–356.
[30] Taichman DB, Backus J, Baethge C, Bauchner H, de Leeuw PW, Drazen JM, et al. Sharing clinical trial data: a proposal from the international committee of medical journal editors. PLoS Med 2016;13:e1001950.
[31] Drazen JM. Data sharing and the journal. N Engl J Med 2016;374:e24.
[32] McNutt M. Data sharing. Science 2016;351:1007.
[33] Wald N. Sharing clinical trial data. J Med Screen 2017;24(2):57.
[34] Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276:637–639.
[35] Novack GD. The CONSORT statement for publication of controlled clinical trials. Ocul Surf 2004;2(1):45–46.
[36] Ellenberg SS, Fleming TR, DeMets DL. Data monitoring committees in clinical trials: a practical perspective. Chichester, West Sussex, England: John Wiley & Sons, Ltd; 2002.
[37] Novack GD. Data monitoring Committees. Ocul Surf 2010;8(1):40–43.
[38] Novack GD. Why aren't there more pharmacotherapies for dry eye? Ocul Surf 2014;12(3):227–230.
[39] Chao W, Belmonte C, Benitez Del Castillo JM, Bron AJ, Dua HS, Nichols KK, et al. Report of the inaugural meeting of the TFOS i(2) = initiating innovation series: targeting the unmet need for dry eye treatment. Ocul Surf 2016;14(2):264–316.
[40] Lemp MA, Bron AJ, Baudouin C, Benítez Del Castillo JM, Geffen D, Tauber J, et al. Tear osmolarity in the diagnosis and management of dry eye disease. Am J Ophthalmol 2011;151(5):792–798. e1.
[41] Stern ME, Beuerman RW, Fox RI, Gao J, Mircheff AK, Pflugfelder SC. The pathology of dry eye: the interaction between the ocular surface and lacrimal glands. Cornea 1998;17(6):584–589.
[42] Stapleton F, Alves M, Bunya VY, Jalbert I, Lekhanont K, Malet F, et al. TFOS DEWS II Epidemiology report. Ocul Surf 2017;15:334–365.
[43] Bron AJ, de Paiva CS, Chauhan SK, Bonini S, Gabison EE, Jain S, et al. TFOS DEWS II Pathophysiology report. Ocul Surf 2017;15:438–510.
[44] Fonn D, Toit R, Simpson TL, Vega JA, Situ P, Chalmers RL. Sympathetic swelling response of the control eye to soft lenses in the other eye. Invest Ophthalmol Vis Sci 1999;40:3116–3121.
[45] Mgarrech M, Rousseau A, Sauer A, Kaswin G, Barreau E, Bourcier T, et al. Lacrymal secretion in the non-affected fellow eye of patients with recurrent unilateral herpetic keratitis. Acta Ophthalmol 2012;90.
[46] Simard-Lebrun A, Boisjoly H, Al-Saadi A, Choremis J, Mabon M, Chagnon M. Association between unilateral quiescent stromal herpetic keratitis and bilateral dry eyes. Cornea 2010;29(11):1291–1295.
[47] Jabbarvand M, Hashemian H, Khodaparast M, Rafatnejad A, Beheshtnejad A, Salami A. Do unilateral herpetic stromal keratitis and neurotrophic ulcers cause bilateral dry eye? Cornea 2015;34:768–772.
[48] Cruzat A, Schrems WA, Schrems-Hoesl LM, Cavalcanti BM, Baniasadi N, Witkin D, et al. Contralateral clinically unaffected eyes of patients with unilateral infectious keratitis demonstrate a sympathetic immune response. Invest Ophthalmol Vis Sci 2015;56(11):6612–6620.
[49] Urzua CA, Vasquez DH, Huidobro A, Hernandez H, Alfaro J. Randomized double-blind clinical trial of autologous serum versus artificial tears in dry eye syndrome. Curr Eye Res 2012;37(8):684–688.
[50] Lemp MA, Sullivan BD. Bilaterality in dry eye disease: implications for clinical trials. In: S DA, editor. 8th international conference on the tear film & ocular surface: basic science and clinical relevance. Montpelier, France; 2016. Abstract #45.
[51] Sullivan BD, Crews LA, Messmer EM, Foulks GN, Nichols KK, Baenninger P, et al. Correlations between commonly used objective signs and symptoms for the diagnosis of dry eye disease: clinical implications. Acta Ophthalmol 2014;92(2):161–166.
[52] Bron AJ, Tomlinson A, Foulks GN, Pepose JS, Baudouin C, Geerling G, et al. Rethinking dry eye disease: a perspective on clinical implications. Ocul Surf 2014;12(2 Suppl):S1–S31.
[53] Sullivan BD, Pepose JS, Foulks GN. Progressively increased variation in tear osmolarity mirrors dry eye severity. JAMA Ophthalmol 2015;133(12):1481–1482.
[54] Pepose Jay S, Sullivan Benjamin D, Foulks Gary N, Lemp Michael A. The value of tear osmolarity as a metric in evaluating the response to dry eye therapy in the clinic and in clinical trials. Am J Ophthalmol 2014;157(1):4–6.
[55] Ederer F. Shall we count numbers of eyes or numbers of subjects? Arch Ophthalmol 1973;89(1):1–2.
[56] Wolffsohn JS, Arita R, Chalmers R, Djalilian A, Dogru M, Dumbleton K, et al. TFOS DEWS II Diagnostic Methodology report. Ocul Surf 2017;15:539–574.
[57] Gold H, Kwit NT. The xanthines (theobromine and aminophylline) in the treatment of cardiac pain. JAMA 1937;108:2173–2179.
[58] Reidenberg MM. Clinical pharmacology: the scientific basis of therapeutics. Clin Pharmacol Ther 1999;66(1):2–8.
[59] Foulks GN. Challenges and pitfalls in clinical trials of treatments for dry eye. Ocul Surf 2003;1(1):20–30.
[60] Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology 2000;107(4):631–639.
[61] Kinoshita S, Oshiden K, Awamura S, Suzuki H, Nakamichi N, Yokoi N, et al. A randomized, multicenter phase 3 study comparing 2% rebamipide (OPC-12759) with 0.1% sodium hyaluronate in the treatment of dry eye. Ophthalmology 2013;120(6):1158–1165.
[62] Tauber J, Karpecki P, Latkany R, Luchs J, Martel J, Sall K, et al. Lifitegrast ophthalmic solution 5.0% versus placebo for treatment of dry eye disease: results of the randomized phase III OPUS-2 study. Ophthalmology 2015;122(12):2423–2431.
[63] Tomlinson A, Hair M, McFadyen A. Statistical approaches to assessing single and multiple outcome measures in dry eye therapy and diagnosis. Ocul Surf 2013;11(4):267–284.
[64] Alves M, Fonseca EC, Alves MF, Malki LT, Arruda GV, Reinach PS, et al. Dry eye disease treatment: a systematic review of published trials and a critical appraisal of therapeutic strategies. Ocul Surf 2013;11(3):181–192.
[65] Novack GD, Crockett RS. Regression to the mean. Ocul Surf 2009;7(3):163–165.
[66] McDonald CJ, Mazzuca SA, McCabe Jr. GP. How much of the placebo 'effect' is really statistical regression? Stat Med 1983 Oct-Dec;2(4):417–427.
[67] Amparo F, Schaumberg DA, Dana R. Comparison of two questionnaires for dry eye symptom assessment: the ocular surface disease index and the symptom assessment in dry eye. Ophthalmology 2015;122(7):1498–1503.
[68] Barton S. Which clinical studies provide the best evidence? The best RCT still trumps the best observational study. Br Med J 2000;321(7256):255–256.
[69] Pinto-Fraga J, López-Miguel A, González-García MJ, Fernández I, López-de-la-Rosa A, Enríquez-de-Salamanca A, et al. Topical fluorometholone protects the ocular surface of dry eye patients from desiccating stress. Ophthalmology 2016;123(1):141–153.
[70] Kangari H, Eftekhari MH, Sardari S, Hashemi H, Salamzadeh J, Ghassemi-Broumand M, et al. Short-term consumption of oral omega-3 and dry eye syndrome. Ophthalmology 2013;120(11):2191–2196.
[71] Hróbjartsson A, Gøtzsche PC. Placebo interventions for all clinical conditions. Cochrane Database Syst Rev 2010;(1):CD003974.
[72] Foulks GN. The evolving treatment of dry eye. Ophthalmol Clin N Am 2003;16(1):29–35.
[73] Novack GD, David R, Lee PF, Freeman MI, Duzman E, Batoosingh AL. Effect of changing medication regimens in glaucoma patients. Ophthalmologica 1988;196(1):23–28.
[74] Maier NRF, Verser GC. Psychology in industrial organizations. 5 ed. Boston: Houghton Mifflin Co;; 1982.
[75] Nelson JD, Gordon JF. Topical fibronectin in the treatment of keratoconjunctivitis sicca. Am J Ophthalmol 1992;114(4):441–447.
[76] Matsumoto Y, Ohashi Y, Watanabe H, Tsubota K, ; Diquafosol Ophthalmic Solution Phase 2 Study Group.. Efficacy and safety of diquafosol ophthalmic solution in patients with dry eye syndrome: a Japanese phase 2 clinical trial. Ophthalmology 2012;119(10):1954–1960.
[77] Sosne G, Ousler GW. Thymosin beta 4 ophthalmic solution for dry eye: a randomized, placebo-controlled, Phase II clinical trial conducted using the controlled adverse environment (CAE™) model. Clin Ophthalmol 2015;9:877–884.
[78] Sosne G, Dunn SP, Kim C. Thymosin B4 significantly improves signs and symptoms of severe dry eye in a phase 2 randomized trial. Cornea 2015:34.
[79] Skelly AC, Dettori JR, Brodt ED. Assessing bias: the importance of considering confounding. Evid Based Spine Care J 2012;3(1):9–12.
[80] Lemp MA. Report of the national eye institute/industry workshop on clinical trials in dry eyes. CLAO J 1995;21(4):221–232.
[81] Eisner A. Sex, eyes, and vision: male/female distinctions in ophthalmic disorders. Curr Eye Res 2015;40:96–101.
[82] Schaumberg DA, Dana R, Buring JE, Sullivan DA. Prevalence of dry eye disease among US men: estimates from the Physicians' Health Studies. Arch Ophthalmol 2009;127(6):763–768.
[83] McMonnies CW. Incomplete blinking: exposure keratopathy, lid wiper epitheliopathy, dry eye, refractive surgery, and dry contact lenses. Contact Lens Anter Eye 2007;30(1):37–51.
[84] Foulks GN, Borchman D, Yappert M. Modification of meibomian gland lipids by topical azithromycin. Invest Ophthalmol Vis Sci 2009;50. ARVO E-Abstract 2676.
[85] Schaumberg DA, Nichols JJ, Papas EB, Tong L, Uchino M, Nichols KK. The international workshop on meibomian gland dysfunction: report of the subcommittee on the epidemiology of, and associated risk factors for, MGD. Invest Ophthalmol Vis Sci 2011;52(4):1994–2005.
[86] Carracedo G, Recchioni A, Alejandre-Alba N, Martin-Gil A, Crooke A, Morote IJ, et al. Signs and symptoms of dry eye in keratoconus patients: a pilot study. Curr Eye Res 2015;40(11):1088–1094.
[87] Gallar J, Acosta MC, Moilanen JA, Holopainen JM, Belmonte C, Tervo TM. Recovery of corneal sensitivity to mechanical and chemical stimulation after laser in situ keratomileusis. J Refract Surg 2004;20(3):229–235.
[88] Kheirkhah A, Dohlman TH, Amparo F, Arnoldner MA, Jamali A, Hamrah P, et al. Effects of corneal nerve density on the response to treatment in dry eye disease. Ophthalmology 2015;122(4):662–668.
[89] Narayanan S, Redfern RL, Miller WL, Nichols KK, McDermott AM. Dry eye disease and microbial keratitis: is there a connection? Ocul Surf 2013;11(2):75–92.
[90] Quinto GG, Campos M, Behrens A. Autologous serum for ocular surface diseases. Arq Bras Oftalmol 2008 Nov-Dec;71(6 Suppl):47–54.
[91] Gomes JAP, Azar DT, Baudouin C, Efron N, Hirayama M, Horwath-Winter J, et al. TFOS DEWS II Iatrogenic report. Ocul Surf 2017;15:511–538.
[92] Rohit A, Willcox MD, Brown SH, Mitchell TW, Stapleton F. Clinical and biochemical tear lipid parameters in contact lens wearers. Optom Vis Sci 2014;91(12):1384–1390.
[93] Brissette AR, Mednick ZD, Schweitzer KD, Bona MD, Baxter SA. Punctal plug retention rates for the treatment of moderate to severe dry eye: a randomized, double-masked, controlled clinical trial. Am J Ophthalmol 2015;160(2):238–242.
[94] Dumbleton K, Caffery B, Dogru M, Hickson-Curran S, Kern J, Kojima T, et al. The TFOS international workshop on contact lens discomfort: report of the subcommittee on Epidemiology. Invest Ophthalmol Vis Sci 2013;54:TFOS20–TFOS36.
[95] Blackwell B. Treatment adherence. Br J Psychiatry 1976;129:513–531.
[96] Blaschke TF, Osterberg L, Vrijens B, Urquhart J. Adherence to medications: insights arising from studies on the unreliable link between prescribed and actual drug dosing histories. Ann Rev Pharm Toxicol 2011;52:275–301.
[97] Stone JL, Robin AL, Novack GD, Covert DW, Cagle GD. An objective evaluation of eyedrop instillation in patients with glaucoma. Arch Ophthalmol 2009;127(6):732–736.
[98] Novack GD. Pipeline: thoughts generated by the annual meeting of the american society of clinical pharmacology and therapeutics. Ocul Surf 2004;2:212–214.
[99] Robin AL, Novack GD, Covert DW, Crockett RS, Marcic TS. Adherence in glaucoma: objective measurements of once-daily and adjunctive medication use. Am J Ophthalmol 2007;144(4):533–540.
[100] Karanicolas PJ, Farrokhyar F, Bhandari M. Practical tips for surgical research: blinding: who, what, when, why, how? Can J Surg 2010;53(5):345–348.
[101] Standardisation IOf. Clinical investigation of medical devices for human subjects - good clinical practice. Int Organ Stand 2011.
[102] Marinho DR, Burmann TG, Kwitko S. Labial salivary gland transplantation for severe dry eye due to chemical burns and Stevens-Johnson syndrome. Ophthalmic Plast Reconstr Surg 2010 May-Jun;26(3):182–184.
[103] Koffler BH, McDonald M, Nelinson DS, Group LACS. Improved signs, symptoms, and quality of life associated with dry eye syndrome: hydroxypropyl cellulose ophthalmic insert patient registry. Eye Contact Lens 2010;36:170–176.
[104] Pult H, Riede-Pult BH, Purslow C. A comparison of an eyelid-warming device to traditional compress therapy. Optom Vis Sci 2012;89:E1035–E1041.
[105] Purslow C. Evaluation of the ocular tolerance of a novel eyelid-warming device used for meibomian gland dysfunction. Contact Lens Anter Eye 2013;36(5):226–231.
[106] Benitez Del Castillo JM, Kaercher T, Mansour K, Wylegala E, Dua H. Evaluation of the efficacy, safety, and acceptability of an eyelid warming device for the treatment of meibomian gland dysfunction. Clin Ophthalmol 2014;8:2019–2027.
[107] Finis D, König C, Hayajneh J, Borrelli M, Schrader S, Geerling G. Six-month effects of a thermodynamic treatment for MGD and implications of meibomian gland atrophy. Cornea 2014;33(12):1265–1270.
[108] Sim HS, Petznick A, Barbier S, Tan JH, Acharya UR, Yeo S, ; Collaborative Research Initiative for Meibomian Gland Dysfunction (CORIM)., et al. A randomized, controlled treatment trial of eyelid-warming therapies in meibomian gland dysfunction. Ophthalmol Ther 2014;3(1–2):37–48.
[109] Choi W, Kim JC, Kim WS, Oh HJ, Yang JM, Lee JB, et al. Clinical effect of antioxidant glasses containing extracts of medicinal plants in patients with dry eye disease: a multi-center, prospective, randomized, double-blind, placebo-controlled trial. PloS One 2015;10(10):e0139761.
[110] Murakami DK, Blackie CA, Korb DR. All warm compresses are not equally efficacious. Optom Vis Sci 2015;92:e327–e333.
[111] Toyos R, McGill W, Briscoe D. Intense pulsed light treatment for dry eye disease due to meibomian gland dysfunction; a 3-year retrospective study. Photomed Laser Surg 2015;33(1):41–46.
[112] Villani Edoardo, Garoli Elena, Canton Veronica, Pichi Francesco, Nucci Paolo, Ratiglia Roberto. Evaluation of a novel eyelid-warming device in meibomian gland dysfunction unresponsive to traditional warm compress treatment: an in vivo confocal study. Int Ophthalmol 2015;35(3):319–323.
[113] Wang MT, Jaitley Z, Lord SM, Craig JP. Comparison of self-applied heat therapy for meibomian gland dysfunction. Optom Vis Sci 2015;92(9):e321–e326.
[114] Wang MT, Gokul A, Craig JP. Temperature profiles of patient-applied eyelid warming therapies. Contact Lens Anter Eye 2015;38(6):430–434.
[115] Greiner JV. Long-term (3 Year) effects of a single thermal pulsation system treatment on meibomian gland function and dry eye symptoms. Eye Contact Lens 2016;42(2):99–107.
[116] Zhao Y, Veerappan A, Yeo S, Rooney DM, Acharya RU, Tan JH, et al. Clinical trial of thermal pulsation (LipiFlow) in meibomian gland dysfunction with preteatment meibography. Eye Contact Lens 2016;42(6):339–346.
[117] Friedman NJ, Butron K, Robledo N, Loudin J, Baba SN, Chayet A. A nonrandomized, open-label study to evaluate the effect of nasal stimulation on tear production in subjects with dry eye disease. Clin Ophthalmol 2016;10:795–804.
[118] Novack GD. The development of drugs vs devices. Ocul Surf 2011;9(1):56–57.
[119] Copay AG, Subach BR, Glassman SD, Polly Jr. DW, Schuler TC. Understanding the minimum clinically important difference: a review of concepts and methods. Spine J 2007 Sep-Oct;7(5):541–546.
[120] Jaeschke R, Singer J, Guyatt GH. Measurement of health status. Ascertaining the minimal clinically important difference. Control Clin Trials 1989;10(4):407–415.
[121] Stratford PW, Binkley JM, Riddle DL, Guyatt GH. Sensitivity to change of the Roland-morris back pain questionnaire: part 1. Phys Ther 1998;78(11):1186–1196.
[122] Nichols KK, Nichols JJ, Mitchell GL. The lack of association between signs and symptoms in patients with dry eye disease. Cornea 2004;23(8):762–770.
[123] Behrens A, Doyle JJ, Stern L, Chuck RS, McDonnell PJ, Azar DT, et al. Dysfunctional tear syndrome: a Delphi approach to treatment recommendations. Cornea 2006;25(8):900–907.
[124] The definition and classification of dry eye disease: report of the definition and classification subcommittee of the international dry eye WorkShop. Ocul Surf 2007;2007(5):75–92.
[125] Sullivan B. Challenges in using signs and symptoms to evaluate new biomarkers of dry eye disease. Ocul Surf 2014;12(1):2–9.
[126] Suzuki M, Massingale ML, Ye F, Godbold J, Elfassy T, Vallabhajosyula M, et al. Tear osmolarity as a biomarker for dry eye disease severity. Invest Ophthalmol Vis Sci 2010;51(9):4557–4561.
[127] Huang JF, Yafawi R, Zhang M, McDowell M, Rittenhouse KD, Sace F, et al. Immunomodulatory effect of the topical ophthalmic Janus kinase inhibitor tofacitinib (CP-690,550) in patients with dry eye disease. Ophthalmology 2012;119(7):e43–e50.
[128] See C, Bilonick RA, Feuer W, Galor A. Comparison of two methods for composite score generation in dry eye syndrome. Invest Ophthalmol Vis Sci 2013;54(9):6280–6286.
[129] Sullivan BD, Eldridge DC, Berg M, Kosheleff V, Porreco A, Truitt J, et al. Diagnostic performance of osmolarity combined with subset markers of dry eye disease in an unstratified patient population. Invest Ophthalmol Vis Sci 2010;51. ARVO E-Abstract 3380.
[130] Leonardi A, Van Setten G, Amrane M, Ismail D, Garrigue JS, Figueiredo FC, et al. Efficacy and safety of 0.1% cyclosporine A cationic emulsion in the treatment of severe dry eye disease: a multicenter randomized trial. Eur J Ophthalmol 2016;26(4):287–296.
[131] Willcox MDP, Argüeso P, Georgiev GA, Holopainen JM, Laurie GW, Millar TJ, et al. TFOS DEWS II Tear Film report. Ocul Surf 2017;15:366–403.
[132] Jackson DC, Zeng W, Wong CY, Mifsud EJ, Williamson NA, Ang CS, et al. Tear interferon-gamma as a biomarker for evaporative dry eye disease. Invest Ophthalmol Vis Sci 2016;57(11):4824–4830.
[133] Baudouin C, Aragona P, Van Setten G, Rolando M, Irkeç M, Benítez del Castillo J, et al. Diagnosing the severity of dry eye: a clear and practical algorithm. Br J Ophthalmol 2014;98(9):1168–1176.
[134] Cardona G, Marcellán C, Fornieles A, Vilaseca M, Quevedo L. Temporal stability in the perception of dry eye ocular discomfort symptoms. Optom Vis Sci 2010;87(12):1023–1029.
[135] Tran N, Graham AD, Lin MC. Ethnic differences in dry eye symptoms: effects of corneal staining and length of contact lens wear. Contact Lens Anter Eye 2013;36(6):281–288.
[136] Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol 2003;136(2):318–326.
[137] O'Brien PD, Collum LM. Dry eye: diagnosis and current treatment strategies. Curr Allergy Asthma Rep 2004;4(4):314–319.
[138] Miller KL, Walt JG, Mink DR, Satram-Hoang S, Wilson SE, Perry HD, et al. Minimal clinically important difference for the ocular surface disease index. Arch Ophthalmol 2010;128:94–101.
[139] Vitale S, Goodman LA, Reed GF, Smith JA. Comparison of the NEI-VFQ and OSDI questionnaires in patients with Sjögren's syndrome-related dry eye. Health Qual Life Outcomes 2004;2:44.
[140] Begley CG, Caffery B, Chalmers RL, Mitchell GL, Dry Eye Investigation (DREI) Study Group. Use of the dry eye questionnaire to measure symptoms of ocular irritation in patients with aqueous tear deficient dry eye. Cornea 2002;21(7):664–670.
[141] Grubbs Jr. JR, Tolleson-Rinehart S, Huynh K, Davis RM. A review of quality of life measures in dry eye questionnaires. Cornea 2014;33(2):215–218.
[142] Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL. Reliability and validity of the ocular surface disease index. Arch Ophthalmol 2000;118(5):615–621.
[143] Schaumberg DA, Gulati A, Mathers WD, Clinch T, Lemp MA, Nelson JD, et al. Development and validation of a short global dry eye symptom index. Ocul Surf 2007;5(1):50–57.
[144] Ridder 3rd WH, Tomlinson A, Huang JF, Li J. Impaired visual performance in patients with dry eye. Ocul Surf 2011;9(1):42–55.
[145] Rajagopalan K, Abetz L, Mertzanis P, Espindle D, Begley C, Chalmers R, et al. Comparing the discriminative validity of two generic and one disease-specific health-related quality of life measures in a sample of patients with dry eye. Value Health 2005 Mar-Apr;8(2):168–174.
[146] Rolando M, Iester M, Macrí A, Calabria G. Low spatial-contrast sensitivity in dry eyes. Cornea 1998;17(4):376–379.
[147] Puell MC, Benítez-del-Castillo JM, Martínez-de-la-Casa J, Sánchez-Ramos C, Vico E, Pérez-Carrasco MJ, et al. Contrast sensitivity and disability glare in patients with dry eye. Acta Ophthalmol Scand 2006;84(4):527–531.
[148] Huang FC, Tseng SH, Shih MH, Chen FK. Effect of artificial tears on corneal surface regularity, contrast sensitivity, and glare disability in dry eyes. Ophthalmology 2002;109(10):1934–1940.
[149] Ridder 3rd WH, Zhang Y, Huang JF. Evaluation of reading speed and contrast sensitivity in dry eye disease. Optom Vis Sci 2013;90(1):37–44.
[150] Goto E, Yagi Y, Matsumoto Y, Tsubota K. Impaired functional visual acuity of dry eye patients. Am J Ophthalmol 2002;133(2):181–186.
[151] Ishida R, Kojima T, Dogru M, Kaido M, Matsumoto Y, Tanaka M, et al. The application of a new continuous functional visual acuity measurement system in dry eye syndromes. Am J Ophthalmol 2005;139(2):253–258.
[152] Denoyer A, Rabut G, Baudouin C. Tear film aberration dynamics and vision-related quality of life in patients with dry eye disease. Ophthalmology 2012;119(9):1811–1818.
[153] Biomarkers Definitions Working G.. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69:89–95.
[154] Hagan S, Martin E, Enríquez-de-Salamanca A. Tear fluid biomarkers in ocular and systemic disease: potential use for predictive, preventive and personalised medicine. EPMA J 2016;7:15.
[155] Lemp MA, Sullivan BD, Crews LA. Biomarkers in dry eye disease. Eur Ophthalmic Rev 2012;6:157–163.
[156] Zhou L, Beuerman RW, Chan CM, Zhao SZ, Li XR, Yang H, et al. Identification of tear fluid biomarkers in dry eye syndrome using iTRAQ quantitative proteomics. J Proteom Res 2009;8(11):4889–4905.
[157] Massingale ML, Li X, Vallabhajosyula M, Chen D, Wei Y, Asbell PA. Analysis of inflammatory cytokines in the tears of dry eye patients. Cornea 2009;28(9):1023–1027.
[158] Epstein SP, Gadaria-Rathod N, Wei Y, Maguire MG, Asbell PA. HLA-DR expression as a biomarker of inflammation for multicenter clinical trials of ocular surface disease. Exp Eye Res 2013;111:95–104.
[159] Wei Y, Gadaria-Rathod N, Epstein S, Asbell P. Tear cytokine profile as a noninvasive biomarker of inflammation for ocular surface diseases: standard operating procedures. Invest Ophthalmol Vis Sci 2013;54(13):8327–8336.
[160] Paletta Guedes RA, Paletta Guedes VM, Freitas SM, Chaoubah A. Does the type of treatment have an influence on utility values in a glaucoma population? Clin Ophthalmol 2015;9:1645–1650.
[161] Bourcier T, Acosta MC, Borderie V, Borrás F, Gallar J, Bury T, et al. Decreased corneal sensitivity in patients with dry eye. Invest Ophthalmol Vis Sci 2005;46(7):2341–2345.
[162] Belmonte C, Nichols JJ, Cox SM, Brock JA, Begley CG, Bereiter DA, et al. TFOS DEWS II Pain and Sensation report. Ocul Surf 2017;15:404–437.
[163] Rosenthal P, Borsook D. Ocular neuropathic pain. Br J Ophthalmol 2016;100(1):128–134.
[164] Goto T, Zheng X, Okamoto S, Ohashi Y. Tear film stability analysis system: introducing a new application for videokeratography. Cornea 2004;23(8 Suppl):S65–S70.
[165] Potvin R, Makari S, Rapuano CJ. Tear film osmolarity and dry eye disease: a review of the literature. Clin Ophthalmol 2015;9:2039–2047.
[166] Abelson MB, Chambers WA, Smith LM. Conjunctival allergen challenge. A clinical approach to studying allergic conjunctivitis. Arch Ophthalmol 1990;108(1):84–88.
[167] Ousler GW, Gomes PJ, Welch D, Abelson MB. Methodologies for the study of ocular surface disease. Ocul Surf 2005;3(3):143–154.
[168] Kojima T, Matsumoto Y, Ibrahim OM, Wakamatsu TH, Uchino M, Fukagawa K, et al. Effect of controlled adverse chamber environment exposure on tear functions in silicon hydrogel and hydrogel soft contact lens wearers. Invest Ophthalmol Vis Sci 2011;52(12):8811–8817.
[169] Meerovitch K, Torkildsen G, Lonsdale J, Goldfarb H, Lama T, Cumberlidge G, et al. Safety and efficacy of MIM-D3 ophthalmic solutions in a randomized, placebo-controlled Phase 2 clinical trial in patients with dry eye. Clin Ophthalmol 2013;7:1275–1285.
[170] Patane MA, Cohen A, From S, Torkildsen G, Welch D, Ousler 3rd GW. Ocular iontophoresis of EGP-437 (dexamethasone phosphate) in dry eye patients: results of a randomized clinical trial. Clin Ophthalmol 2011;5:633–643.
[171] Semba CP, Torkildsen GL, Lonsdale JD, McLaurin EB, Geffin JA, Mundorf TK, et al. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am J Ophthalmol 2012;153(6):1050–1060. e1.
[172] Novack GD. Translating drugs from animals to humans: do we need to prove efficacy? Transl Vis Sci Technol 2013;2:1–4.
[173] Tang-Liu DD, Liu SS, Weinkam RJ. Ocular and systemic bioavailability of ophthalmic flurbiprofen. J Pharmacokinet Biopharm 1984;12(6):611–626.
[174] Novack GD, Moyer ED. How much nonclinical safety data are required for a clinical study in ophthalmology? J Ocul Pharmacol Ther 2016 Jan-Feb;32(1):5–10.
[175] Edeki TI, He H, Wood AJJ. Pharmacogenetic explanation for excessive b-blockade following timolol eye drops: potential for oral-ophthalmic drug interaction. JAMA 1995;274:1611–1613.
[176] Novack GD, Stamer WD. The “T” in JOPT. J Ocul Pharmacol Ther 2015;31:129.
[177] Haidar SH, Davit B, Chen ML, Conner D, Lee L, Li QH, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res 2008;25(1):237–241.
[178] Davit BM, Nwakama PE, Buehler GJ, Conner DP, Haidar SH, Patel DT, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother 2009;43(10):1583–1597.
[179] Woodcock J. Evidence vs. access: can twenty-first-century drug regulation refine the tradeoffs? Clin Pharmacol Ther 2012;91(3):378–380.
[180] Levitan B. A concise display of multiple end points for benefit-risk assessment. Clin Pharmacol Ther 2011;89(1):56–59.
[181] Poland B, Hodge FL, Khan A, Clemen RT, Wagner JA, Dykstra K, et al. The clinical utility index as a practical multiattribute approach to drug development decisions. Clin Pharmacol Ther 2009;86(1):105–108.
[182] Korsan B, Dykstra K, Pullman W. Transparent trade-offs. Pharm Exec 2005.
[183] Goyal N, Gomeni R. A novel metric to assess the clinical utility of a drug in the presence of efficacy and dropout information. Clin Pharmacol Ther 2012;91(2):215–219.
[184] Novack GD. The benefit/risk of good therapeutics. Ocul Surf 2012;10(4):264–266.
[185] Novack GD. Decoding the packaging insert: indications. Ocul Surf 2003;1(3):150–151.
[186] Gadek TR, Burdick DJ, McDowell RS, Stanley MS, Marsters Jr. JC, Paris KJ, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 2002;295(5557):1086–1089.
[187] Anonymous. Assessment report: Ikervis. 2016.
[188] Kinoshita S, Awamura S, Oshiden K, Nakamichi N, Suzuki H, Yokoi N, et al. Rebamipide (OPC-12759) in the treatment of dry eye: a randomized, double-masked, multicenter, placebo-controlled phase II study. Ophthalmology 2012;119(12):2471–2478.
[189] Kinoshita S, Oshiden K, Awamura S, Suzuki H, Nakamichi N, Yokoi N, ; Rebamipide Ophthalmic Suspension Phase 3 Study Group.. A randomized, multicenter phase 3 study comparing 2% rebamipide (OPC-12759) with 0.1% sodium hyaluronate in the treatment of dry eye. Ophthalmology 2013;120(6):1158–1165.
[190] Greiner JV. A single LipiFlow® Thermal Pulsation System treatment improves meibomian gland function and reduces dry eye symptoms for 9 months. Curr Eye Res 2012;37(4):272–278.
[191] Finis D, Hayajneh J, König C, Borrelli M, Schrader S, Geerling G. Evaluation of an automated thermodynamic treatment (LipiFlow®) system for meibomian gland dysfunction: a prospective, randomized, observer-masked trial. Ocul Surf 2014;12(2):146–154.
[192] Anonymous. De novo classification request for TearScience, Inc. Lipiflox(R) thermal pulsation system. Device Classification under Section 513(a)(1)(de novo). 2010.
[193] Gumus K, Schuetzle KL, Pflugfelder SC. Randomized controlled crossover trial comparing the impact of sham or intranasal tear neurostimulation on conjunctival goblet cell degranulation. Am J Ophthalmol 2017;177:159–168.
[194] 513(f)(2) - Evaluation of automatic class III designation, guidance for industry and CDRH staff. In: Evaluation OoD, editor. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES, FDA, Center for Devices and Radiological Health, Rockville, MD 208501998.
[195] Geerling G, Raus P, Murube J. Minor salivary gland transplantation. Dev Ophthalmol 2008;41:243–254.
[196] Geerling G, Sieg P. Transplantation of the major salivary glands. Dev Ophthalmol 2008;41:255–268.
[197] International Conference on Harmonisation. Statistical principles for clinical trials (E9). 1998.
[198] Novack GD. The Importance of A Priori statistical planning in controlled clinical trials. Am J Ophthalmol 2015;160(1):4–5. e1.
[199] Huque M, Rohmel J. Multiplicity problems in clinical trials: a regulatory perspective. In: Dmitrienko A, Tamhane AC, Bretz F, editors. Multiple testing problems in pharmaceutical statistics. Boca Raton, FL: CRC Press/Taylor & Francis; 2010. p. 1–33.
[200] Musch DC, Gillespie BW. The state of being noninferior. Ophthalmology 2006;113(1):1–2.
[201] Novack GD. Some are more equal than others. Ocul Surf 2014;12(2):155–158.
[202] Novack GD. Ophthalmic beta-blockers since timolol. Surv Ophthalmol 1987 Mar-Apr;31(5):307–327.
[203] Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, et al. Treatment of dry eye syndrome with orally administered CF101: data from a phase 2 clinical trial. Ophthalmology 2010;117(7):1287–1293.
[204] Capita L, Chalita MR, dos Santos-Neto LL. Prospective evaluation of hypromellose 2% for punctal occlusion in patients with dry eye. Cornea 2015;34(2):188–192.
[205] Simmons PA, Carlisle-Wilcox C, Chen R, Liu H, Vehige JG. Efficacy, safety, and acceptability of a lipid-based artificial tear formulation: a randomized, controlled, multicenter clinical trial. Clin Ther 2015;37(4):858–868.
[206] Steven P, Scherer D, Krösser S, Beckert M, Cursiefen C, Kaercher T. Semifluorinated alkane eye drops for treatment of dry eye disease-a prospective, multicenter noninterventional study. J Ocul Pharmacol Ther 2015;31(8):498–503.
[207] Sheppard JD, Torkildsen GL, Lonsdale JD, D'Ambrosio Jr. FA, McLaurin EB, Eiferman RA, et al. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology 2014;121(2):475–483.
[208] Shimmura S, Ono M, Shinozaki K, Toda I, Takamura E, Mashima Y, et al. Sodium hyaluronate eyedrops in the treatment of dry eyes. Br J Ophthalmol 1995;79(11):1007–1011.